
アミノ酸関連酵素の立体構造に基づいた基質の認識·触媒機構の研究
博士論文 糖質·アミノ酸関連酵素の立体構造に基づいた基質の認識·触媒機構の研究 平成22年進学 林 到炫 指導教員 伏信進矢 目次 目次 ........................................................................................................................................... 3 略語 ........................................................................................................................................... 3 序章 ........................................................................................................................................... 5 1. 糖質·糖質加水分解酵素について............................................................................... 6 2. アミノ酸·アミノ酸関連酵素について..................................................................... 12 図表 ................................................................................................................................... 17 引用文献 ........................................................................................................................... 30 第1章 X線結晶構造解析によるThermotoga martima MSB8由来 GH51 アラビノフラ ノシダーゼの基質認識触媒機構の解明 ............................................................................. 32 序 ....................................................................................................................................... 33 1.1 Thermotoga martima由来アラビノフラノシダーゼ ........................................ 33 1.2 Glycoside Hydrolaseの新規な活性化誘導体基質としてのDMT糖 ................ 36 実験の目的と方法 ........................................................................................................... 38 1.3 Tm-AFaseの大腸菌を用いた大量発現と精製 ................................................ 39 1.4 Tm-AFaseの結晶化条件の探索 ........................................................................ 41 1.5 X線回折データ測定 .......................................................................................... 42 1.6 分子置換法による初期構造決定及び構造精密化 ......................................... 43 1.7 基質複合体の構造解析 ..................................................................................... 44 1.8 Automated-docking法によるDMT誘導体の触媒様式の予測.......................... 45 結果と考察 ....................................................................................................................... 47 1.9 組み換えTm-AFaseの精製 ................................................................................ 47 1.10 Tm-AFaseの結晶化条件の探索結果 .............................................................. 47 1.11 構造解析の結果 ............................................................................................... 48 1.12 構造に関する考察 ........................................................................................... 49 1.13 本章のまとめ ................................................................................................... 54 図表 ................................................................................................................................... 56 引用文献 ........................................................................................................................... 85 第2章 新規二機能性フラビン酵素Pseudomonas sp. AIU 813由来L-アミノ酸オキシダ ーゼ/オキシゲナーゼの二機能性反応機構に関する構造生物学的研究 ........................ 88 序 ....................................................................................................................................... 89 2.1 Pseudomonas sp. AIU 813由来のL-アミノ酸オキシダーゼ/オキシゲナーゼ.... 89 実験の目的と方法 ........................................................................................................... 92 2.2 Native L-AAOd/Ogの発現と精製 .................................................................... 93 2.3 L-AAOd/Ogセレノメチオニン置換体の作成 ................................................. 95 2.4 Native L-AAOd/OgとL-AAOd/Og Se-Met置換体の結晶化条件探索 ............. 96 2.5 X線回折データ測定 ........................................................................................... 98 1 2.6 初期構造決定及び構造精密化 ......................................................................... 98 2.7 基質複合体の構造解析 ..................................................................................... 99 2.8 変異体酵素C254Iの構造解析 ......................................................................... 100 2.9 pCMB修飾L-AAOd/Ogの構造解析 ................................................................ 101 2.10 L-AAOd/Ogの諸性質の解析 ......................................................................... 102 結果と考察 ..................................................................................................................... 104 2.11 組み換えL-AAOd/Ogの精製と諸性質の解析 ............................................. 104 2.12 L-AAOd/Ogの結晶化条件の探索結果 ......................................................... 105 2.13 構造解析の結果 ............................................................................................. 106 2.14 構造に関する考察 ......................................................................................... 107 2.15 本章のまとめ ................................................................................................. 112 図表 ................................................................................................................................. 115 引用文献 ......................................................................................................................... 144 総括 ....................................................................................................................................... 147 発表論文 ............................................................................................................................... 151 謝辞 ....................................................................................................................................... 152 2 略語 4A·A: 4-Aminoantipyrine AB: o-Aminobenzoate AFase: Arabinofuranosidase BSA: Bovine Serum Albumin CAZy: Carbohydrate Active enZymes CBM: Carbohydrate-Binding Module CE: Carbohydrate Esteraase crLAAO: L-Amino acid oxidase from Calloselasma Rhodostoma D-Xyl: D-Xylose DMT: 4,6-Dimethoxy-1,3,5-triazin-2-yl EB: Emerald BioSystems EG: Ethylene Glycol FAD: Flavin-Adenine Dinucleotide FMO family: Flavin Monooxygenase family FOM: Figure of Merit FPLC: Fast Protein Liquid Chromatograph GH: Glycoside Hydrolase Gs-AFase: α-L-Arabinofuranosidase from Geobacillus stearothermophiles GT: Glycosyl Transferase HR: Hampton Research IPTG: Isopropyl-β-D(-)-thiogalactopyranoside IUBMB: International Union of Biochemistry and Morecular Biology KEK: 高エネルギー加速器研究機構、茨城県つくば市 L-AAOd/Og: L-Lysine-α-oxidase/2-monooxygenase 3 L-Ara: L-Arabinose L-Arg: L-Arginine L-Lys: L-Lysine L-Orn: L-Ornithine LAAO family: L-Amino Acid Oxidase family LB: Luria-Bertani LGA: Lamarckian Genetic Algorithm MAD: Multiple-wavelength Anomalous Dispersion method MIR: Multiple Isomorphous Replacement method MR: Molecular Replacement PAO: Phenylalanine oxidase/monooxygenase from Psuedomonas sp. P-501 pCMB: p-Chloromercuribenzoate PDB: Protein Data Bank PEG: Polyethylene glycol PL: Polysaccharide Lyase PLP: Pyridoxal phosphate pNP: p-Nitrophenyl RMSD: Root Mean Square Deviation SAD: Single-wavelength Anomalous Dispersion SDS-PAGE: SDS ポリアクリルアミド電気泳動 Se-Met: Selenomethionine Spring-8: 高輝度光科学研究センター、兵庫県西播磨 SV-LAAO: L-Amino acid oxdiases from Snake venom Tm-AFase: α-L-Arabinofuranosidase from Thermotoga maritima TOOS: N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3methylaniline sodium salt dehydrate 4 序章 5 1. 糖質·糖質加水分解酵素について 1-1 糖質(Saccharide or Carbohydrate) 糖質はタンパク質、脂質、核酸に並ぶ重要な生体物質の 1 つである。その分子式 は Cm(H2O)n と表される。糖質は基本単位である単糖、それらが 3 つ以上結合したオ リゴ糖、さらに多数の単糖からなる多糖に分けられる。単糖のヘミアセタールと別 の単糖のヒドロキシル基は、脱水縮合反応でアセタールを形成し、この結合様式を グリコシド結合という(図 1(a))。C1 位のアノマー炭素には化学的に異なる四つの置 換基が結合しており、α または β の立体配置を取ることができる。一方、糖質はそ の長さ(Degree of Polymerization)により二糖類、オリゴ糖(DP = 3~20)、多糖類に分け られる(図 1(b, c))。 ・二糖類(Disaccharide) 2 つの単糖がグリコシド結合でつながったものを二糖という。グルコース 2 分子 が α-1,4 結合したマルトース(麦芽糖)、グルコースとフルクトースが α-1,4 結合した スクロース(ショ糖)、ガラクトースとグルコースが β-1,4 結合したラクトース(乳糖) などが代表的な二糖である。 ・オリゴ糖(Oligosaccharide) 3 ~ 20 分子ほどの単糖がつながったものをオリゴ糖という。ラフィノース、ゲン チアノースなどが代表的なオリゴ糖である。人の母乳の成分にも様々なオリゴ糖(ヒ トミルクオリゴ糖、HMO)が含まれているのが明らかになっており、これに関する 研究が活発に行われている。 ・多糖類(Polysaccharide) 単糖類が多数グリコシド結合したものを多糖類という。同一種の単糖類から成る ものをホモ多糖、種以上の単糖類から成るものをヘテロ多糖という。主な多糖類で はデンプンやセルロースなどがある。 1-2 糖質の役割 6 糖質の機能としては、大まかに分けて以下の 3 種類があると言われている。 ・エネルギー源 ほとんどの生物は解糖によるグルコースの分解により、エネルギー通貨としての ATP などの高エネルギー化合物を合成する。Embden-Meyerhof-Parnas 解糖系はグル コースを分解してピルビン酸にまで変換する過程で、2 分子の ATP が合成される。 また、低分子化合物からエネルギーの貯蔵のためにグルコース、さらにデンプンや グリコーゲンなどの高分子をつくり出す糖新生も行われている。 ・構造糖質 セルロースは細胞の骨格を構成する成分である。細胞壁に含まれるセルロースの 基本構造は Glcβ1-4Glc の繰り返しであり、その構造は直鎖状である。水酸基が分子 間・分子内水素結合を形成することでセルロースが水との接触を欠く不溶性繊維とし て機能する。その他にも、マンノースを主成分とするマンナン、カニの甲殻や昆虫 の外骨格の主成分であるキチンやキトサンなどがある。 ・複合糖鎖 さまざまな単糖が組み合わさってできた糖鎖、さらにそれがタンパク質や脂質と 結合した「複合糖鎖」はその結合様式により N-結合型糖鎖と O-結合型糖鎖に分けられ る。これらは、ABO 式血液型に見られるように、各種抗原決定基群を形成するほか、 糖鎖を認識するタンパク質であるレクチンとの相互作用を通じて、輪送·接着·シグ ナル伝達などの細胞間相互作用や、細胞の環境因子認識において重要な役割を持っ ていることが知られている。最近は医療分野を中心に、糖鎖の機能に注目した薬品· 食品が開発されている。 1-3 糖質関連酵素 多様な形で存在する糖質には作用する酵素の数も非常にたくさんある。糖質の分 解や転移などに関わる酵素は糖質関連酵素と呼ばれる。糖質関連酵素のデータベー ス CAZy (Carbohydrate-Active Enzymes; 後述)では、作用機構に基づいて Glycoside Hydrolase (GH, 糖質加水分解酵素, EC3.2.1.-)、Glycosyl Transferase (GT, 糖転移酵素, EC2.4.-.-)、Polysaccharide Lyase (PL, 多糖リアーゼ, EC4.2.2.-)、Carbohydrate Esterase 7 (CE, 炭水化物エステラーゼ)、Carbohydrate-Binding Module (CBM, 糖質結合モジュ ール)の 4 つのクラスに分類している。各クラスの作用機構を図 2 に示す。 GH は主に糖質加水分解酵素を含む。糖質加水分解酵素とは、糖と糖間、糖と非 糖間の O-グリコシド(または S-グリコシド)の加水分解を触媒する酵素である。GH ファミリーではその他に、糖転位反応の活性が高いトランスグリコシダーゼや、加 リン酸分解反応を行うホスホリラーゼなどが含まれている。2013 年現在、125 ファ ミリーが設立されており、CAZy の約半分を占めている最大のクラスである[1]。 また、糖質関連酵素は、触媒ドメインの前後に比較的小さな非触媒の糖質結合ド メインを持っていることがある。これらは酵素ではないが基質を認識して結合する 働きをしており、CBM として現在 64 ファミリーが設立されている。 本研究の第 1 章では、これらの分類の中で 糖質加水分解酵素 GH51 ファミリーに 属する酵素を取り扱った。 1-4 GH ファミリーの分類 糖質加水分解酵素の IUBMB (International Union of Biochemistry and Molecular Biology)による酵素の命名法はそれらの基質特異性や分子機構に基づいているが、こ れらの酵素に関する構造上の特徴は反映していない。B. Henrissat らはアミノ酸配列 と立体構造・反応機構には直接的な因果関係があると考え、アミノ酸相同性に基づい てファミリーに分類した[2, 3](表 1)。これらの分類はウェブ上でデータベース化され ており、CAZy (Carbohydrate‐Active enZYmes、http://www.cazy.org/)で取りまとめられ ている。この分類法により同一ファミリーに属している糖質加水分解酵素は、以下 のような特徴があると考えられている。 ·同一ファミリーに属している酵素は、類似の立体構造を持つ。 ·同一ファミリーに属している酵素は、進化的に関連がある。 ·同一ファミリーに属している酵素は、同一の反応機構(1-5 節)を示す。 GH ファミリーは、GH1 から GH131 まで分類されていて糖質関連酵素の中で最大 のクラスである(2013 年 1 月現在)。このうち、GH21、GH40、GH41、GH60、GH69、 GH91 は、一度ファミリーに分類化されたが、他のファミリーとの関連が明らかに なり再分類されたことから、欠番となっている(表 2)。また、GH の中で、同じフォ 8 ールドを持ち、触媒残基が同じ位置に存在する比較的近縁なファミリーは Clan とし てまとめられている(図 3)、(表 3)。 1-5 GH の反応機構 糖質加水分解酵素はその加水分解の立体化学的結果に基づいて、アノマー保持型 (retaining)もしくはアノマー反転型(inverting)に分類される[4, 5]。その活性中心はほ とんどの場合 2 つの酸性残基(アスパラギン酸またはグルタミン酸)であり、その反 応機構は一部の例外を除いて以下の 2 種類に分けられる。 ・アノマー保持型(Retaining mechanism) アノマー保持型反応機構は、2 つのステップで反応を触媒する。それぞれのステ ップでアノマー炭素の結合を反転させるため、結果的にもとの構造を保持する。反 応には 2 つの酸性残基が、1 つは求核性触媒残基、もう 1 つは酸/塩基触媒残基とし て働く。最初のステップで求核性触媒残基はアノマー中心部に求核攻撃し、酸/塩基 触媒残基がプロトンを供与する。この時、求核性触媒残基と糖が共有結合を形成し、 アシル中間体を生成する。次のステップでは、プロトンを奪われた酸/塩基触媒残基 が塩基として水分子からプロトンを引き抜き、この水が中間体を求核攻撃すること で加水分解する反応が起こる(図 4(a))。この反応の第 2 段階において、水分子以外の 分子が求核剤として働くと糖の転移反応が起こる。基質自身が求核剤として働くこ とで基質の重合反応が起こる。第 1 章の DMT-Xyl による糖転移反応は、このような 反応で起こる(第 1 章)。 上述のように、アノマー保持型の反応機構は、求核剤による攻撃が 2 回起こるた め、double displacement mechanism と呼ばれる。二つの触媒残基環距離は酸/塩基触媒 -基質-求核性触媒となるため約 5.5Åである。 第 1 章で対象とした GH51 アラビノフラノシダーゼは典型的なアノマー保持型機 構に従い、活性中心に、求核性触媒(nucleophile)および酸/塩基触媒(acid/base catalyst) として 2 つのグルタミンが存在する[6]。 ・アノマー反転型(Inverting mechanism) アノマー反転型反応機構は、2 つの酸性残基が 1 組となって触媒を行う。それぞ れ一般塩基触媒、一般酸触媒残基として働く。グリコシド結合に対し、一般酸触媒 がプロトンを供与し、一般塩基触媒による脱プロトン化で活性化した水分子が求核 9 攻撃する。その時、オキソカルボニウムイオンと呼ばれる中間体を経て、アノマー が反転する(図 4(b))。アノマー反転型反応機構は、求核攻撃が 1 回のみ起こることに より、Single displacement mechanism と呼ばれる。二つの触媒残基間距離は一般酸触 媒-基質-水(求核剤)-一般塩基性触媒となるため約 6.5 ~ 9.5 Å である。 1-6 糖質分解酵素の活性部位 一般的に酵素分子には、われ目やくぼみなどが存在する。基質が酵素に結合する と、この部分にはまり込み、反応が起こる。このような場所を活性部位あるいは活 性中心と呼ぶ。 ・結合のサブサイト 酵素反応が起こるためには、基質(糖)が酵素の結合部位に正しく結合することが 必要である。一般的に多糖の分解酵素は、複数個所の糖鎖構造を認識する活性中心 部位を持っている。糖質加水分解酵素において、このような基質の認識部位をサブ サイトと表現する。グリコシド結合の切断が起こる部位を基準:0 として、還元末端 側の構成糖結合部位をサブサイト+1、+2、+3・・・として表し、非還元末端側をサ ブサイト-1、-2、-3・・・と表す(図 5)。 ・エンド型加水分解酵素とエキソ型加水分解酵素 糖質加水分解酵素は、糖鎖のどの部分に作用するかによってエンド型とエキソ型 に分類される。エンド型加水分解酵素は、糖鎖にランダムに作用して加水分解する。 エキソ型加水分解酵素は、糖鎖の非還元末端に作用して順番に糖を切り出す酵素で ある。例えば、エンドグルカナーゼはセルロース鎖をランダムに切断するエンド型 加水分解酵素であり、セロビオヒドロラーゼはセルロースの非還元末端からセロビ オース単位(2 糖)ずつ切断するエキソ型加水分解酵素である。 一般的に、糖質加水分解酵素の基質結合サブサイトは、その-1 位が注目されて いる。触媒反応における糖のコンフォメーション変化はサブサイト-1 位で起こる 上に、酵素の命名法もサブサイト-1 位がどのような糖質構造を認識するかによっ て決まる。たとえば、第 1 章の研究対象であるアラビノフラノシダーゼは、サブサ イト-1 位がアラビノースを認識するため、アラビノフラノシダーゼという名前が 付けられた。これまで糖質関連酵素研究の多くは基質結合サブサイト-1 のみに注 10 目して行われている。しかし、酵素の生体内の役割を考える上で、+側のサブサイト の基質特異性は非常に重要な情報である。また糖質関連酵素を糖質合成へ利用する 蛋白質工学において、+側サブサイトの認識の構造情報は、難合成オリゴ糖や有用オ リゴ糖を高収率で合成するためには不可欠である(第 1 章)。 本研究では、その構造が未知であるエンド型グリコシダーゼ GH51 ファミリーに 属する好熱菌由来のアラビノフラノシダーゼ(第 1 章)を対象とし、サブサイト+1 位 の結合様式が実際の酵素反応にどのような影響を及ぼすかを明らかにすることを目 的として立体構造解析を目指した。 11 2. アミノ酸·アミノ酸関連酵素について 2-1 アミノ酸(Amino acid) アミノ酸は主に生体タンパク質の構成ユニットであるが、天然に産する広義のア ミノ酸の中には、旨み成分や、薬物として作用するもの、そして毒となるものなど も存在している。アミノ酸はアミノ基(-NH2)とカルボキシル基(-COOH)を炭素骨格 にもつ化合物である。アミノ酸には非常に多くの種類があるが、タンパク質の材料 となるものは 20 種に限られる(表 4)。カルボキシル基が結合する炭素を α 炭素(Cα、 不剤炭素原子)といい、この Cα に結合したカルボキシル基、アミノ基、水素以外の 残りの 1 箇所に結合するものを各アミノ酸特有の原子団あるいは側鎖(-R)という。 アミノ酸には L 型と D 型の光学異性体が存在するが(図 6(a))、タンパク質を構成す るアミノ酸はすべて L-α-アミノ酸である。一方、最初のアミノ酸のカルボキシル基 の OH と 2 番目のアミノ酸のアミノ基の H が脱水縮合(水分子が 1 個取れる形で 2 個 の分子が結合する)するペプチド結合(図 6(b))によりペプチド鎖が伸長し、オリゴペ プチド、ポリペプチドを形成する。 ・アミノ酸の分類 タンパク質をつくる 20 種類のアミノ酸を、側鎖(あるいは、-R)の種類により分類 することができる(表 4)。親水性アミノ酸はセリン、トレオニン、アスパラギン、グ ルタミンなどで、疎水性アミノ酸はアラニン、イソオイシン、ロイシン、メチオニ ン、フェニルアラニン、トリプトファン、バリン、チロシンなどである。アミノ酸 の電気的性質により正に荷電している塩基性アミノ酸のリジン、アルギニン、ヒス チジンと、負に荷電する酸性アミノ酸のグルタミン酸、アスパラギン酸に分けられ る。特別なアミノ酸として、システインはスルフヒドリル基-SH を含み、2 つの-SH 間で S-S 結合(ジスルフィド結合)を形成する。グリシンは-R が水素であるため最も 簡単な構造を取っており、プロリンは α 炭素と窒素が環状構造に入る特徴がある。 別の分け方としては脂肪族(炭素と水素を基本とした非環状構造)の側鎖を持つもの と、芳香族(ベンゼン環構造)の側鎖を持つ芳香族アミノ酸などがある。このよなア ミノ酸の性質はタンパク質の性質にも反映されるため、非常に重要である。 2-2 アミノ酸代謝とアミノ酸関連酵素 アミノ酸は単に組織タンパク質の構成成分として必要であるばかりではなく、そ の炭素骨格は糖や脂質の材料としてエネルギー生産に用いられる。またアミノ基は 12 様々な含窒素化合物の合成に用いられて、ビタミン、ホルモン、生体色素の形成に も関係するほか、核酸塩基の重要な構成要素となっている。したがってアミノ酸代 謝は生体内で極めて多様で重要な機能をしている。さらに、このようなアミノ酸代 謝の異常は、先天性アミノ酸代謝症候群から見られているため、これらに関する研 究の重要性はより大きくなっている。(アミノ酸代謝異常については後節で詳しく説 明する) 一方、アミノ酸関連酵素のほとんどはアミノ酸代謝過程に関わる酵素である。本 章の 1-3 で紹介した糖質関連酵素は、CAZy というデータベースを中心とし、アミノ 酸配列、立体構造及び触媒反応機構により明確に分類されているが、アミノ酸関連 酵素は広範囲である酵素の種類や機能のため、酵素全般に関しての分類体系が明確 に構築されてない。しかし、アミノ酸関連酵素については昔から研究されており、 各酵素の触媒反応機構、特に各酵素によるアミノ酸の代謝機構などが詳しく知られ ている。ここでは、アミノ酸の代謝と共にその過程で作用するアミノ酸関連酵素を 紹介する。 2-3 アミノ酸の窒素の代謝 ・酸化的脱アミノ反応 L-アミノ酸酸化酵素(L-amino acid oxidase, EC1.4.3.2)、D-アミノ酸酸化酵素(Damino acid oxidase, EC1.4.3.3)は、それぞれ L-および D-アミノ酸の酸化的脱アミノ反 応(oxidative deamination)を触媒し、対応するケト酸(オキソ酸)を生成する酵素である [7]。これらの酵素はフラビンアデニンジヌクレオチド(FAD)を補酵素とし、アミノ 酸の分解利用の役割を持つと考えられており、生成したケト酸からの ATP 合成を可 能とする。本論文の第 2 章の対象酵素である L-アミノ酸オキシダーゼ/オキシゲナー ゼは 2 つの反応を触媒するが、そのうち 1 つはこの L-アミノ酸酸化酵素と同じ反応 を触媒する。 アミノ酸脱水素酵素(amino acid dehydrogenase, EC1.4.1.5)は、NAD+、NADP+を補酵 素として、L-アミノ酸の可逆的脱水素反応を触媒する。特に、生理的に有意義な酵 素としてグルタミン酸脱水素酵素(glutamate dehydrogenase, EC1.4.1.2)があり、アミノ 酸代謝と有機酸代謝の転換点の重要な役割を果たしている[8]。また、フェニルアラ ニ ン に 高 い 基 質 特 異 性 を 示 す フ ェ ニ ル ア ラ ニ ン 脱 水 素 酵 素 (phenylalanine dehydrogenase, EC1.4.1.20)は昔から研究されており、フェニルアラニンケトン尿症の 診断キットとして実用化されている[9]。 ・アミノ基転移反応 13 アミノ基転移反応を触媒する酵素群は、補酵素としてビタミン B6 であるピリド キサールリン酸(pyridoxal phosphate, PLP)を持つ。アミノ基転移酵素(amino transferase, transaminase)は、アミノ酸とケト酸の相互交換を触媒し、アミノ酸代謝の中心的存在 である。 アミノ基転移反応は、特定のアミノ酸の α-アミノ基を酵素中の PLP に転 移し、ピリドキサミン-5’-リン酸(PMP)と基質である特定のアミノ酸に対応するケト 酸を生成する。次に、α-ケトグルタル酸は PMP からアミノ基を受け取り、PLP を再 生し、グルタミン酸を生成する。これらの 2 つの半反応は平衡反応であり、全反応 も平衡反応である。代表的なアミノ基転移酵素としては、アスパラギン酸トランス フェラーゼ(aspartate transaminase, EC2.6.1.1)がある[10]。 ・脱水的脱アミノ反応 セリンとトレオニンは共に、脱水反応を伴った脱アミノ反応より分解を受ける。 この反応は、PLP を補酵素とする酵素により触媒される。代表的な酵素としてはセ リンデヒトラターゼ(serine dehydratase, EC4.3.1.17)がある[11]。 ・ラセミ化反応 アミノ酸のラセミ化を触媒するアミノ酸ラセマーゼ(amino acid racemase)の多くは、 アミノトランスフェラーゼと同様に PLP を補酵素とする酵素である[12]。可逆的に L-アミノ酸を D-アミノ酸に変換する反応を触媒する。グリシン、セリン、トレオニ ンの代謝、システイン代謝、D-グルタミンおよび D-グルタミン酸代謝、D-アルギニ ン及び D-オルニチン代謝の4つの代謝に関与する。 2-4 アミノ酸代謝異常 Archibald Garrod (1857–1936)は、遺伝性疾患の原因が代謝酵素の欠損であることを 明らかとして、遺伝子と酵素の関連を最初に示すとともに、先天性代謝異常(inborn errors of metabolism)の概念を提唱した。表 5 にヒトの先天性アミノ酸代謝異常につ いて示した。代表的なアミノ酸代謝異常症を以下で論じる。 ・フェニルケトン尿症 フェニルケトン尿症(phenylketonuria, PKU)は、一原子酸素添加酵素であるフェニ ルアラニン水酸化酵素(phenylalanine hydroxylase)の欠損が病因である。フェニルアラ ニンをチロシンへ変換できないため、フェニルアラニンの血中の値が上昇する。こ 14 のためフェニルアラニンが通常は起こらない代謝を受け、アミノ基転移反応の結果、 フェニルピルビン酸(フェニルケトン)が大量に作られ、さらにその代謝産物である フェニル乳酸やフェニル酢酸とともに、尿中に大量に配泄される。この症状は、出 生時には確かめられず、乳児期後半から中枢神経系異常を示すようになり、重度の 神経遅滞となる。 ・アルカプトン尿症(チロシン代謝) アルカプトン尿症(alkaptonuria)は、チロシン代謝中間体であるホモゲンチジン酸 の酸化酵素(二原子酸素添加酵素)の欠損で起こる。ホモゲンチジン酸が蓄積して、 尿中に多量に排出される。 その他にも、チロシナーゼの欠損から起因する色素欠乏症(白皮症、albinism)、ホ モシスチンとセリンからシスタチオニンを合成する酵素の欠損によるホモシスチン 尿症(homocystinuria)、分岐 α-ケト酸脱水素酵素縮合体の欠損による、メープルシロ ップ尿症(maple syrup urine disease)などがアミノ酸代謝異常症として知られている。 最近は、アミノ酸関連酵素を用いてこれらの先天性代謝異常を検出するアミノ酸定 量キットの開発が活発に行われている。 2-5 アミノ酸オキシダーゼとフラビンモノオキシゲナーゼ 前述のように、L-アミノ酸オキシダーゼ(L-amino acid oxidase, LAAO)は L-アミノ 酸の酸化的脱アミノ反応を触媒して α-ケト酸、アンモニア、過酸化水素を生成する FAD 依存性の酵素である(図 7(a))。現在まで、カビ、藻類、バクテリアなどの様々 な生物種からの LAAO が発見されているが、そのうちもっとも昔から研究されてき たのが蛇毒由来の L-アミノ酸オキシダーゼ(SV-LAAO)である[13]。LAAO はその基 質特異性により、大きく 2 つのグループに分けられる。1 つあるいはわずかなアミ ノ酸を基質とする酵素群には L-グルタミン酸オキシダーゼ(EC1.4.3.11)、L-リジンオ キシダーゼ(EC1.4.3.14)、L-アスパラギン酸オキシダーゼ(EC1.4.3.16)、L-フェニルア ラニンオキシダーゼ(EC1.13.12.9)などがあり、他のグループは様々なアミノ酸を基 質とする基質特異性が広い酵素群である。 一方、フラビンモノオキシゲナーゼ(Flavin monooxygenase, FMO, EC 1.13 and 1.14) は二原子酸素のうちの 1 原子がヒドロキシ基として基質に取り込まれ、他の 1 原子 は水に還元される反応を触媒する酵素である(図 7(b))。この酵素は沢山の反応を触媒 する幅広いファミリーであり、芳香環ヒドロキシ化反応、酸化的脱炭酸反応、及び 15 アミンの酸化などを行う。この酵素の反応は、補酵素の FAD がアミノ酸基質のアミ ノ基と反応してイミノ基と還元状態の FAD になるところから始まる。 電子豊富な FAD 中間体は酸素分子を基質として使用し、1 つの電子が還元された FAD から酸素 に転移されてスーパーオキシドとフラビンラジカルを形成する。ほとんどの FMO はフラビンの C4a と酸素分子が結合して安定的な C4a-ヒドロペルオキシフラビン (hydroperoxyflavin)を形成する。ペルオキシフラビンは不安定であり、すぐペルオキ シドや酸化型フラビンを生成するが、ここで FMO は基質を酸素化することで安定 化させることができる。これらの結果、酸素分子からの 1 つの原子は水に還元され る[14]。巨大ファミリーである FMO は、その触媒反応様式、基質の種類、立体構造 のトポロジーに基づいてクラス A からクラス F まで 6 つのファミリーに分類されて いる(表 6)[15]。 本論文の第 2 章で取り扱ったターゲット酵素、L-アミノ酸オキシダーゼ/オキシゲ ナーゼは、上述した 2 つの酵素(アミノ酸オキシダーゼ及びフラビンモノオキシダー ゼ)の特徴を同時に持つ二機能性酵素である。両酵素は異なる触媒反応機構を有して いるが、FAD を補酵素として用いることや触媒反応中に酸素が重要な役割をするこ となど共通点も存在している。両酵素の反応機構に関しては、Pseudomonas sp. P-501 由来の L-フェニルアラニンオキシダーゼ(PAO)でその立体構造に基づいた研究がな されている[16]。PAO はその基質の 1 つである L-フェニルアラニンに対してはオキ シゲナーゼ反応を、L-メチオニンに対してはオキシダーゼ反応を触媒する特徴があ る。本研究の対象酵素は、L-リジンに対してオキシダーゼ及びオキシゲナーゼとし て働く酵素であり、まだその立体構造が未知である。また、p-クロロ水銀安息香酸 (pCMB) 処理により両活性のうちオキシゲナーゼ活性が低下してオキシダーゼ活性 が 5 倍以上昇することが最近の研究により明らかになっている(第 2 章)。 本研究の第 2 章では、構造未知の L-アミノ酸オキシダーゼ/オキシゲナーゼの立体 構造を明らかにし、両方の触媒反応機構を明らかにすることを試みた。また、 pCMB による活性の転移メカニズムを生物構造学的な観点から確立するのを目的と してこの研究を行った。 16 図表 触媒能による分類 (Class) 立体構造に基づく分類 (Clan) 一次構造に基づく分類 (Family) 糖質加水分解酵素 (GH, Glycoside Hydrolase) GH-A ~ GH-N GH1 ~ GH131 糖転移酵素 (GT, Glycosyl Transferase) GT-A ~ GT-B GT1 ~ GT94 多糖リアーゼ (PL, Polysaccharide Lyase) - PL1 ~ PL22 炭水化物エステラーゼ (CE, Carbohydrate Esterase) - CE1 ~ CE16 糖質結合ドメイン (CBM, Carbohydrate-Binding Module) fold1 ~ fold7 CBM1 ~ CBM64 (2013 年 1 月現在) 表 1 糖質関連酵素の分類 17 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 22 23 24 25 26 27 28 29 30 32 33 34 35 36 37 38 39 42 43 44 45 46 47 48 49 51 52 53 54 55 56 57 58 59 61 62 63 64 65 66 67 68 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 31 50 70 131 (2013 年 1 月現在) **欠番になっているのは、他のクラスに再分類されたファミリーなどである。青色は立体構造が明らかになったファミリー、未 知のファミリーは灰色に示した。黄色に示したファミリーは、本研究で取り扱った酵素が属しているファミリーである。その立 体構造は解明されている。 表 2 GH ファミリーの分類 18 Clan Fold Family GH-A (β/α)8 1, 2, 5, 10, 17, 26, 30, 35, 39, 42, 50, 51, 53, 59, 72, 79, 86, 113, 128 GH-B β-jelly roll 7, 16 GH-C β-jelly roll 11, 12 GH-D (β/α)8 27, 31, 36 GH-E 6-fold β-propeller 33, 34, 83, 93 GH-F 5-fold β-propeller 43, 62 GH-G (α/β)6 37, 63 GH-H (β/α)8 13, 70, 77 GH-I α+β 24, 46, 80 GH-J 5-fold β-propeller 32, 68 GH-K (β/α)8 18, 20, 85 GH-L (α/α)6 15, 65, 125 GH-M (α/α)6 8, 48 GH-N β-helix 28, 49 (2013 年 1 月現在) 表 3 GH ファミリーの Clan 分類 19 3文字 字 表記 記 1文字 表記 グリシン Gly y G ヒスチジン Hiss H リジ ジン Lyss K アル ルギニン Arg g R 酸 アスパラギン酸 p Asp D NH -CH2N H -(CH2)4-N NH3 -(CH2)3-N NH-C=NH2 NH2 -CH2-COO O- グル ルタミン酸 Glu u E -CH2-CH2-COO - アスパラギン Asn n N -CH2-CO-N NH2 グル ルタミン Gln n Q -CH2-CH2-CO-NH 2 セリン Serr S -CH2OH トレ レオニン Thrr T -CH(OH)-C CH3 ニン フェニルアラニ Phee F -CH2- チロシン Tyrr Y -CH2- トリプトファン ン Trp p W メチ チオニン Mett M N H -CH2-CH2-S-CH-CH 3 システイン Cyss C -CH2-SH アラニン Alaa A -CH3 ロイシン Leu u L イソロイシン Ile I バリン Vall V プロリン Pro o P 性質 名称 中性 正電荷をも もつ 親水性 負電荷をも もつ アミドを含 含む ヒドロキシ基 を含む 芳香環をも もつ 疎水性 硫黄を含む む 脂肪族の性質 をもつ 側鎖 鎖の構造 -H -CH2- CH H3 -CH2-CH CH H3 CH2 CH H3 -CH CH3 CH -CH 3 CH3 NH 表 4 タンパク質を タ をつくる 20 2 種類のア アミノ酸 20 OH CO OOH 病患名 欠損経路 欠損酵素 フェニルケトン尿症 フェニルアラニンから チロシンへの変換 アルカプトン尿症 チロシンの分解 色素欠乏症(白皮症) チロシンからのメラニ ン合成 ホモシスチン尿症 メチオニンの分解 フェニルアラニンヒド ロキシラーゼ ホモゲンチジン酸 1 ,2- ジ オ キ シ ゲ ナ ー ゼ チロシン 3-モノオキ シゲナーゼ(チロシナ ーゼ) シスタチオン β-シン ターゼ メープルシロップ尿 症 (分岐ケト酸尿症) イソロイシン、ロイシ ン、バリンの分解 分枝鎖の α-ケト酸デ ヒドロゲナーゼ複合体 嘔吐、痙攣、 精神発達遅滞、早期死亡 メチルマロン酸血症 プロピオニル CoA か らスクシニル CoA へ の変換 メチルマロニル CoA ムターゼ 嘔吐、痙攣、 精神発達遅滞、早期死亡 高グリシン血症 グリシンの分解 グリシン開裂系酵素複 合体 アルギニン血症 尿色合成 シトルリン血症 尿色合成 カルバモイルリン酸 シンテターゼⅠ欠損 症 尿色合成 アルギナーゼ アルギニノコハク酸シ ンテターゼ カルバモイルリン酸シ ンテターゼⅠ 表 5 ヒトの先天性アミノ酸代謝異常 21 症状及び結末 新生児嘔吐、精神発達遅滞 黒色尿、遅発性関節炎 色素欠乏、白髪、白色皮膚 骨発育不良、精神発達遅滞 痙攣、呼吸障害などの精神 症状、早期死亡、精神発達 遅滞 精神発達遅滞 嘔吐、痙攣 嗜眠、痙攣、早期死亡 Subclass A Prototype 4-OH-benzoate hydroxylase B Cyclohexanone monooxygenase C Luciferase D 4-OH-phenylacetate hydroxylase E Styrene monooxygenase F Tryptophane 7-halogenase Structure(PDB code); fold Hydroxylation epoxidation α FAD NAD(P)H 2 (1PHH, 1FOH); 1 FAD/NAD(P)binding domain Baeyer-Villiger; N-oxidation α FAD NADPH 214/309 1 (1W4X); 2 FAD/NAD(P)binding domains, 1 helical domain Light emission; S-oxidation; α + β FMN NAD(P)H 432/309 2 (1BRL, 1M41); Baeyer-Villiger TIM barrel Hydroxylation α+β FAD NAD(P)H 43/309 0; Acyl-CoA dehydrogenase Epoxidation α+β FAD NAD(P)H 21/309 0; 1 FAD/NAD(P)binding domain Halogenation α+β FAD NAD(P)H 102/309 1 (2APG); 1 FAD/NAD(P)binding domain, 1 helical domain 「W.J.H. van Berkel et al., Journal of Biotechnology, 2006. 124, 670–689」より抜粋 Reactions Subunits Cofactor Coenzyme 表 6 フラビンモノオキシゲナーゼ(FMO)の分類 22 Genome frequency 570/309 図 1 糖質の種 種類 a) グリ リコシド結合 合の形成 b) 自然 然界に存在す する主な二 二糖類 c) 多糖 糖類 (デンプ プンやセル ルロース) 23 図 2 各クラスの の糖質関連 連酵素の作用 用機構 質加水分解酵 酵素(GH): 糖-糖間 のグリコシ シド結合、または糖- -非糖間の のグリコ a) 糖質 シド結 結合を加水分 分解する。 b) 糖転 転移酵素(G GT): 糖供与 与体の糖を特 特異的な糖 糖受容体に転移し、新 新たなグリコシド 結合を作り出す。 c) 多糖 糖リアーゼ((CE): 糖鎖 鎖を β-脱離 離機構で分解 解し、新た たにできた還 還元末端に に二重結 合を導 導入する。 d) 糖エ エステラーゼ ゼ(PL): O--アセチル基 基、まだは は N-アセチ チル基を脱離 離する反応 応を触媒 する。 24 図 3 GH の多様な なクランと とそのフォー ールド 25 図 4 糖質加水 分解酵素(G GH)の反応 応機構 me (アシル ル保持型酵素 素) a) Retaiining Enzym ま ず Poton doonor で あ る 一 般 酸 / 塩 基 触 媒 が プ ロ ト ン を 供 給 し 、 そ れ に 共 に Nucleopphile(求核性 性触媒残基 基)もグリコ コシド結合を を攻撃して て求核置換反 反応を起こ こす。次 に、Caatalytic base により活 活性化され れた水分子が が同じ位置 置の炭素に求 求核置換反 反応を起 こす。このように に 2 段階の の反応が起 起こることで で、アノマ マーが保持さ される反応 応を保持 型反応 応という。 b) Inverrting Enzym me (アシル反 反転型酵素 素) Protoon donor(一 一般酸触媒))の Proton 供 供給と同時 時に、Cataly ytic base(一 一般塩基触媒)によ り活性 性化された水 水分子がグ グリコシド結 結合を攻撃 撃して求核置換反応を を起こす。このよ うに段 段階の反応を を起こすことで、アノ ノマーが反 反転する反応 応を反転型 型反応という う。 26 図 5 酵素 素の結合サ サブサイト a) 酵素 素の結合サブ ブサイトで での基質の結 結合様式 b) アラ ラビノフラノシダーゼ ゼのサブサイ イトの構造 造 Geobaccillus stearoothermophilus 由来ア アラビノフラノシダー ーゼと基質 4-nitropheenyl-Ara の結合 合様式。アラ ラビノフラノース部分 分がサブサ サイト-1 に結合するの に のが見られ れる。 「Hovel, K., et al., Thhe EMBO jo ournal, 2003. 22(19), 4922-32」より抜 抜粋 27 図 6 アミノ酸 酸の構造と とぺプチド結 結合 ミノ酸が持つ つ 2 種類の の光学異性体 体: L-アミノ酸(左)、D-アミノ酸 酸(右) a) アミ b) アミ ミノ酸のペプチド結合 合 28 図 7 L-アミノ酸オキ キシダーゼ ゼ及びモノオ オキシゲナ ナーゼの触媒 媒反応機構 構 a) L-アミノ酸オキ キダーゼ(L LAAO)の触 触媒反応機構 構。この反 反応によって て α-ケト酸 酸、アン モニア、過酸化水 水素が生成 成される。 「Ibrahim m M. Moustaafa et al., J. Mol. M Biol., 20006. 364, 991–1002」より り抜粋 b) フラ ラビンモノオ オキシゲナ ナーゼ(FMO O)の触媒反 反応機構。二 二原子酸素 素中に 1 原子は基 原 質のヒドロキシ基 基に取り込 込まれ、もう う 1 個の酸 酸素原子は は水として還 還元される る。この 図では、NAD(P)H H 依存性の の FMO の反 反応機構を を示す。 「W.J.H. van Berkel et e al., Journaal of Biotechn hnology, 2006 6. 124, 670–6 689」より抜 抜粋 29 引用文献 1. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V & Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource f or Glycogenomics. Nucleic Acids Res 37, D233-238. 2. Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280 (Pt 2), 309-316. 3. Henrissat B & Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293 (Pt 3), 78 1-788. 4. Davies G & Henrissat B (1995) Structures and mechanisms of glycosyl hydrolas es. Structure 3, 853-859. 5. McCarter JD & Withers SG (1994) Mechanisms of enzymatic glycoside hydrolys is. Curr Opin Struct Biol 4, 885-892. 6. Hovel K, Shallom D, Niefind K, Belakhov V, Shoham G, Baasov T, Shoham Y & Schomburg D (2003) Crystal structure and snapshots along the reaction path way of a family 51 alpha-L-arabinofuranosidase. Embo J 22, 4922-4932. 7. Bender AE & Krebs HA (1950) The oxidation of various synthetic alpha-aminoacids by mammalian d-amino-acid oxidase, l-amino-acid oxidase of cobra venom and the l- and d-amino-acid oxidases of Neurospora crassa. Biochem J 46, 210219. 8. Lightfoot DA, Baron AJ & Wootton JC (1988) Expression of the Escherichia-Co li Glutamate-Dehydrogenase Gene in the Cyanobacterium Synechococcus Pcc6301 Causes Ammonium Tolerance. Plant Mol Biol 11, 335-344. 9. Tachibana S, Suzuki M & Asano Y (2006) Application of an enzyme chip to th e microquantification of l-phenylalanine. Anal Biochem 359, 72-78. 10. Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN, Gehring H & Christ en P (1984) Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J Mol Biol 174, 497-525. 11. Selim AS & Greenberg DM (1959) An enzyme that synthesizes cystathionine an d deaminates L-serine. J Biol Chem 234, 1474-1480. 12. Soda K & Osumi T (1969) Crystalline amino acid racemase with low substrate specificity. Biochem Biophys Res Commun 35, 363-368. 13. Massey V & Curti B (1967) On the reaction mechanism of Crotalus adamanteus L-amino acid oxidase. J Biol Chem 242, 1259-1264. 14. van Berkel WJ, Kamerbeek NM & Fraaije MW (2006) Flavoprotein monooxyge nases, a diverse class of oxidative biocatalysts. J Biotechnol 124, 670-689. 30 15. Massey V (2000) The chemical and biological versatility of riboflavin. Biochem Soc Trans 28, 283-296. 16. Ida K, Suguro M & Suzuki H (2011) High resolution X-ray crystal structures of L-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas s p. P-501. Structures of the enzyme-ligand complex and catalytic mechanism. J B iochem 150, 659-669. 31 第1章 X 線結晶構造解析による Thermotoga martima MSB8 由来 GH51 アラビノフラノシダーゼの基質認識触媒機構の解明 (Crystallography of Glyscoside Hydrolase Family 51 α-L-arabinofuranosidase from Thermotoga maritima MSB8) 32 序 1.1 Thermotoga maritima 由来アラビノフラノシダーゼ ・植物細胞壁構成成分とその分解酵素 植物細胞壁のリグノセルロース(lignocellulose)は、セルロース、ヘミセルロース、 ペクチン、リグニンで構成されている(図 1-1)。セルロースは、D-グルコピラノース が β-1,4 結合で重合したものであり、比較的単純な構造をとっている。分子間水素結 合を形成していることでセルロース 36 ~ 50 本がまとまって、セルロース微小繊維を 構成する[1]。ヘミセルロースは、セルロースとペクチン系多糖を除く陸上植物細胞 壁を構成する多糖類と定義されており、地球上で最も豊富な再生可能なポリマーの 一つである。その構造は、キシラン、マンナンなどを主鎖とし、ガラクトースやア ラビノースが側鎖として結合している(図 1-2)[2]。植物細胞の構成量は植物の種類に よって多様である(表 1-1)。セルロース、ヘミセルロース、ペクチン、リグニンは、 リグノセルロースの分解において鍵となるコンポーネントであり、これらの重合体 基質の分解過程には様々な酵素が関わっている[3]。 ヘミセルロース分解酵素 ヘミセルロースの 1 種であるアラビノキシランは、D-キシロースが β-1,4 結合で 重合しているキシラン主鎖に、側鎖として L-アラビノース、D-ガラクトース、アセ チル基、フェルロ酸、p-クマル酸、グルクロン酸などの多様な側鎖が置換されてい る複雑な構造をとっている(図 1-2)。そのため、ヘミセルロースを分解する酵素の種 類は非常に多い[4]。ここでは、アラビノキシラン分解に関わる酵素について述べる (図 1-3)。キシランの主鎖分解にはキシラナーゼ、β-キシロシダーゼが関与する。キ シラナーゼ(EC 3.2.1.8)は、キシランの β-1,4 結合をランダムに加水分解するエンド型 の加水分解酵素であり、キシラン分解の主役である[5]。β-キシロシダーゼ(EC 3.2.1.37)は、キシランやキリロオリゴ糖の β-1,4 結合を非還元末端から分解するエキ ソ型加水分解酵素でキシロースを遊離する。 アラビノキシランは図 1-2、図 1-3 のようにアラビノースなどの側鎖が結合した側 鎖構造を持つが、キシラナーゼ、キシロシダーゼが働くためにはまずこれらの側鎖 を分解する必要がある。図 1-3 にアラビノキシランの側鎖に結合している様々な修 飾基を加水分解する酵素を記す。これらは、アクセサリー酵素と呼ばれており[6]、 アラビノフラノース側鎖を切断するアラビノフラノシダーゼ(EC 3.2.1.55)、グルクロ 33 ン酸側鎖を切断するグルクロニダーゼ(EC 3.2.1.139)、側鎖のアラビノースに結合し たフェルラ酸を切断するフェルラ酸エステラーゼ(EC 3.1.1.73)、アセチル基を切断す るアセチルキシランエステラーゼ(EC 3.1.1.72)などがある。 ・アラビノフラノシダーゼ L-アラビノシル残基は、アラビナン、アラビノキシラン、アラビアガム、アラビ ノガラクタンなどのヘミセルロースに多く分布している。アラビノフラノシダーゼ (α-L-arabinofuranosidase、AFase)は、これらの基質に含まれる α-1,2、α-1,3、α-1,5 結 合で結合した非還元末端のアラビノース(アラビノフラノース)をエキソ型で切断す るアクセサリー酵素である[7]。以下にはアラビノフラノシダーゼの分類及び有用性 について述べる。 アラビノフラノシダーゼは、触媒する基質の種類、反応機構などの特徴から様々 な種類が存在する。アミノ酸配列による CAZy の GH family の分類でも、沢山のフ ァミリーに分類されている(表 1-2)。GH3 は幅広い基質特異性を示すファミリーであ り数多くの酵素が属されている。このファミリーに属する α-L-アラビノフラノシダ ーゼは β-キシロシダーゼの活性を同時に持つ特徴がある。GH43 の酵素群も幅広い 基質特異性を持っており、α-L-アラビノフラノシダーゼ、アラビナナーゼ、β-キシ ロシダーゼなどが属する。このファミリーはアノマー反転型の触媒機構を取ってい るのが特徴であり、現在まで 3 種類の AFase の立体構造が報告されている。GH51 には α-L-アラビノフラノシダーゼ、エンドグルガナーゼが属している。本研究の対 象酵素はこのファミリーに分類される。GH51 AFase については、後で詳しく説明す る。GH54 には、主に α-L-アラビノフラノシダーゼが分類されているが、キシロシ ダーゼ活性を持つものを 1 つ含んでいる。Aspergillus 属や Fusarium 属などカビ由来 の AFase のみがこのファミリーに分類される。アノマー保持型の触媒機構を取って おり、現在まで 1 種類の立体構造が当研究室より解明されている[8]。GH62 には、 α-L-アラビノフラノシダーゼのみが分類されている。Aspergillus 属などのカビや Streptomyces 属などの放線菌由来の AFase が分類される。このファミリーの AFase は、可溶性の基質を加水分解できず、不溶性の基質しか加水分解できないことが報 告されている。CBM2、CBM13、CBM35 といったキシラン結合能を持つ CBM を有 しているのが特徴である。反応機構は明らかになっておらず、構造未知のファミリ ーである。最近の研究により新たな AFase が見つかり、新しい GH ファミリーに分 類された。GH121、GH127 に分類された β-アラビノフラノシダーゼは、従前の AFase とは異なって β-結合からなるアラビノフラノオリゴ糖鎖を加水分解する酵素 であり、Bifidobacterium 属由来のものが報告されている。これらの立体構造につい ては、現在まで未発表である。 34 ヘミセルロースの 1 つであるアラビノキシランは、側鎖にアラビノースが結合し ているため分解が進みにくく、あまり有効利用されてこなかった。そこで、アラビ ノフラノシダーゼで側鎖のアラビノースを取り除いてやることにより、その後キシ ラナーゼによる分解が飛躍的に進む。分解された糖は、バイオエタノールへのアル コール発酵や乳酸発酵の基質として利用することができる。また、単糖としての Lアラビノースは、小腸粘膜に存在するスクラーゼの働きを特異的に阻害するなどの 機能性を持つことで注目されている。砂糖(スクロース)と一緒に摂取することで砂 糖の消化や吸収を抑制し、血糖値の急激な上昇を抑制することができる。このよう に、L-アラビノースは、機能性食品素材として注目されており、アラビノフラノシ ダーゼを用いた L-アラビノースの生産が期待される。 ・Thermotoga maritima 由来 α-L-アラビノフラノシダーゼ T. maritima は、無胞子形成の Thermotogales 目に属する高度好熱菌真正細菌であり、 イタリアの海底火山付近の地熱で温められた海底の堆積物から単離されたものであ る[9]。その至適生育温度は 80°C である。T. maritima は small subunit ribosomal RNA による進化系統樹において、進化の速度が遅いとされる最も枝の深いところに位置 する真正細菌の一種であるため、生物の進化を考える上で重要な生物であるといえ る[10]。ゲノムプロジェクトによって全塩基配列が解明されていおり、グルコース、 スクロース、デンプン、セルロース、ヘミセルロースを代謝する酵素を持っている [11-13]。ヘミセルロース代謝酵素としては、これまでに 2 種類のエンドキシラナー ゼ[14]、キシロシダーゼ[15]、2 種類のグルクロニダーゼ[16]の機能解析が行われて いる。近年、アラビノフラノシダーゼ(Tm-AFase)がクローニングされてその機能解 析が行われた[17]。Tm-AFase はアミノ酸配列から GH51 に分類される(表 1-2)。TmAFase と最も高いアミノ酸配列の相同性(配列同一性 36%)を有する GH51 酵素として は、Geobacillus stearothermophilus 由来アラビノフラノシダーゼ(Gs-AFase)の立体構 造が解析されている[18]。Gs-AFase は Retaining 酵素であり、その触媒残基が明らか になっている。Tm-AFase では acid/base catalyst と nucleophile が、各々Glu172、 Glu281 であり(図 1-4)、変異体解析からこれらの残基が活性に非常に重要な残基であ ることが確認されている。組換え Tm-AFase (55 kDa)は、p-nitrophenyl(pNP)-α-Larabinofuranoside に強い活性を示すが(Km = 0.416 mM, kcat= 21.7 s-1, kcat/Km = 5.22×104 M-1s-1)、pNP-β-D-xylopyranoside に対しても kcat/Km の比で約 1/1000 の微弱な活性を示 す(Km = 2.02 mM, kcat = 0.116 s-1, kcat/Km = 57.3 M-1s-1)。これはアラビノフラノシダーゼ には一般的に見られる性質で、アラビノフラノースとキシロピラノースが構造的に 似ていることに起因すると考えられている。また、Tm-AFase は超好熱性かつ耐熱性 酵素であり、その至適温度は 90°C である。90°C で 24 時間インキュベートしても失 活せず、100°C で 20 分間インキュベートすると 50%まで活性が落ちるが、その後半 35 減期 2.7 時間でゆっくり失活する(図 1-5)[17]。したがって、Tm-AFase は現在まで知 られているアラビノフラノシダーゼの中で最も耐熱性の酵素である。 1.2 Glycoside Hydrolase (GH)の新規な活性化誘導体基質としての DMT 糖 GH の合成基質としては、pNP-, o-nitrophenyl-, dinitrophenyl-などのニトロフェニル 基を導入した配糖体が古くから用いられてきた。しかし、近年、東北大学の正田晋 一郎教授らのグループにより、4,6-dimethoxy-1,3,5-triazin-2-yl (DMT)環を導入した配 糖体の合成法と利用法の開発が進められている。DMT 誘導体は以下のような特徴を 持つ。 a) 合成が pNP に比べて圧倒的に簡単であること。pNP では有機溶媒中で少なくとも 4 段階は必要だが、DMT では水中で一段階で合成可能である。 b) GH の基質として非常に高い一般性を持つ。正田らのグループではこれまでに約 10 種類の DMT 誘導体を合成しているが、全て、関連する酵素で加水分解されると いう結果が得られている。 c) pNP と同程度、あるいは酵素によっては pNP よりも高い加水分解活性を示す。 d) 特定の酵素との組み合わせにより、DMT 誘導体は高い糖転移活性を示し、多糖 合成反応のドナーとして用いられることが分かっている[19]。 本研究では特に上記の b-c に関する構造的知見を得るために、サブサイト(+1)の DMT 基との相互作用を明らかにすることを目的とし、ここでは Tm-AFase と DMT 基質を研究対象とした。 東北大正田研では、Tm-AFase の基質特異性からアラビナン側鎖だけではなく、キ シラン主鎖にも作用してキシロースを切り出すことに着目し、キシロースに DMT を導入した合成基質 DMT-β-O-xylopyranoside (DMT-Xyl)を用いてキシロオリゴ糖の 合成反応が試みられた。図 1-6 には Tm-AFase とキシロース誘導体(DMT-Xyl と pNPXyl)の酵素反応の結果を表した。Tm-AFase の非存在下では DMT-Xyl がゆっくり加 水分解されるが(lane 2)、DMT-Xyl が Tm-AFase の基質として用いられたときは、反 応後 1 時間でいくつかのスポットが観察された(lane 3)。pNP-Xyl が Tm-AFase と反 応したときは反応後 24 時間目でも生成物がないことが lane 5 から確認された。300 mM Xyl-β-DMT, 0.155 mg/ml AFase, pH 6.0, 30°C の条件で 1 時間反応を行ったときの、 反応産物を HPLC で分離して NMR で各ピークを同定した(図 1-7, 1-8)。その結果、 36 加えた基質の約 20%が重縮合反応を起こし、少なくとも 3 種類のオリゴ糖が生成さ れているのが確認された(図 1-9)。 一般的に好熱菌由来の糖質加水分解酵素は安定で扱いやすく、産業的なスケール で生産することができるといった利点がある。Tm-AFase は、現在まで報告されてい るアラビノフラノシダーゼの中で最も耐熱性があるものであるため、Tm-AFase と DMT-Xyl を組み合わせた糖合成反応は新規のキシロース合成系としてより期待され ている。また、Gs-AFase の例を含めて、pNP 基質と GH 酵素の複合体構造は多数報 告されているものの[18]、DMT 誘導体と GH の複合体の構造はこれまで全く報告が ない。本研究では、DMT 誘導体と Tm-AFase のサブサイト(+1)の相互作用を明らか にするために、立体構造が未知である Tm-AFase の構造決定を目指した。また、 Xyl-β-DMT の O- グ リ コ シ ド 結 合 を S- グ リ コ シ ド 結 合 に 変 え た DMT-β-Sxylopyranoside (Xyl-S-DMT)を用いて、その反応中間体の構造を決定するために TmAFase との共結晶化を試みた。 37 実験の目的と方法 ・試薬 本研究で用いた試薬は特に記さない限り和光純薬工業株式会社、SIGMA-Aldrich 社のものを使用した。 ・実験器具 吸光光度計 V-550 (JASCO)、Benchmark Plus (BIO-RAD)、Nanodrop ND-1000 を活 性測定などに使用した。超音波破砕機は BIORUPTOR UCW-201 型 (COSMO BIO)、 SONIFIER 250D (BRANSON)を使用した。 ・培地 本研究ではすべて大腸菌を用いて実験を行ったため、特に表記しない限り LB (Luria-Bertani)培地(DIFCO)を用いた。すべての LB 培地はオートクレーブ滅菌を使っ た。試験管培養は、水槽式インキュベーター(TAITEC)を使用し、大量培養の場合は、 低温恒温槽付回転式振とう培養機(高杉製作所)を使用した。抗生物質を添加する場 合は、カナマイシン 50 μg/ml を培地に添加して培養を行った。 ・遺伝子操作 Molecular Cloning 及び Current Protocol in Molecular Biology に記されている方法に 従った。 ・発現ベクター·菌株 本研究で取り扱ったアラビノフラノシダーゼは、産業総合研究所·生物プロセス研 究部門·合成生物工学研究グループの宮崎健太郎博士らにより遺伝子がクローニング され、大腸菌による発現ベクターが作成された。提供していただいた発現用のプラ スミドは、pET-28a(+)のマルチクローニングサイトに 5'側は EcoR I で、3’側は Sal I で切り出された Tm-AFase の遺伝子(1452b.p.)を組み込まれたものであり、N 末端に His-Tag が付加されている(図 1-10)。プラスミドの保持用の菌株は E. coli XL10GOLD (Stratagene) を 用 い 、 発 現 用 菌 株 と し て は E. coli BL21 (DE3)-Codon plus 38 (Stratagene)を用いた。この菌株はクロラムフェニコール耐性を持っているため、形 質転換の際には LB プレートにクロラムフェニコールとカナマイシンを各々、100 μg/ml、50 μg/ml となるように添加した。 1.3 Tm-AFase の大腸菌を用いた大量発現と精製 ・目的 本研究の主な目的である X 線構造解析のためにタンパク質の結晶化を行う。良質 の結晶を作るためには、高純度で大量のタンパク質が必要である。そのために、ま ず発現条件の検討を行い、最も大量に発現する条件を見つけ出して大量発現·精製系 の構築を行った。 ・組み換え大腸菌の大量発現 ① E. coli BL21 (DE3)-CodonPlus に発現ベクターpET-28a(+)を形質転換した。 ② ①で作成した組換え大腸菌をグリセロールストックより、LB 培地 10 ml に植菌 し、37°C で一晩振盪培養で前培養を行った。 ③ 前培養した 10 ml の培養液を LB 培地 3 L (5 L 羽根つき三角フラスコに 1.5 L 入 れたものを 2 本)に植菌し、37°C で本培養を開始した。 ④ 約 5 時間後に、終濃度 0.5 mM の IPTG (Isopropyl-β-D(-)-thiogalactopyranoside)を添 加し、同じ温度でさらに約 5 時間振盪培養した。 ⑤ 培養液を回収して 8,000 rpm、10 分間の遠心で集菌した。 ・Tm-AFase の精製 大量培養して集菌した Tm-AFase の菌体を 150 ml の 20 mM Tris-HCl (pH 8.0)緩衝 液で懸濁し、出力レベル 5.5、Duty 50%、Palse 1 sec で、25 分間超音波破砕を行った。 その後、12,000 rpm、45 分間の遠心で集菌した。上清の可溶性画分のみを採取し、 Minisart syringe filter 0.45 μm (Sartorius Stedim)を用いて微粒子を除去した。 熱処理(Heat Treatment) 39 Tm-AFase は好熱性細菌由来の酵素であるため、その精製ステップの一環で熱処理 を行った。大量発現した Tm-AFase の酵素液を 70°C で 30 分間熱処理を行った。熱 処理は水槽式インキュベーターで行った。その後、15,000 rpm、30 分間遠心し、そ の上清の可溶性画分を採取して Minisart syringe filter 0.45 μm を用いて微粒子を除去 した。 Ni-NTA Superflow column chromatography この精製の制御には、ペリスタポンプ(Econo gradient pump, Bio-RAD)を用いた。 ① C 16/20 column (GE Healthcare Bioscience)に Ni-NTA superflow resin (QIAZEN)を 5 ml 充填し、20 mM Tris-HCl (pH 8.0)を用いて平衡化した。 ② サンプルを①に添加し、20 mM Tris-HCl (pH 8.0) + 30 mM Imidazole で非吸着画 分を溶出した。 ③ 20 mM Tris-HCl (pH 8.0) + 100 mM Imidazole を用いて吸着画分を溶出した。 ④ 分離した吸着画分を Amicon Ultra-15 10K (Millipore) を用いて濃縮·脱塩。濃縮脱 塩後のバッファーは 20 mM Tris-HCl (pH 8.0)用いた。 Gel filtration column chromatography ·Running buffer: 20 mM Tris-HCl pH 8.0 + 150 mM NaCl ① Ni カラムクロマトグラフィーで分離した吸着画分は Ultrafree-MC 0.45 μm を用い て微粒子を除去した。 ② Hiload 16/60 Superdex 200 prep grade (GE Healthcare Bioscience)カラムを Running buffer で平衡化した。流速は 1.0 ml/min。 ③ 1 ml 容量のサンプルループを用いてサンプルをカラムに吸着させ、Running buffer で溶出した。 ④ 280 nm の UV モニターリングでタンパク質の分離を確認し、分離した画分を Amicon Ultra-4 10K で濃縮·脱塩を行った。 ・タンパク質定量 40 タンパク質の定量は、BCA (Bicinchoninic acid)法により行った。BCA working reagent (PIERCE)の A と B バッファーを 50:1 で混ぜた溶液とサンプルを 20:1 で混合 し、60°C で 30 分間反応した後、562 nm の吸光を測定した。標準タンパク質として は、BSA (Bovine Serum Albumin)を用いて 0、0.05、0.1、0.15、0.2、0.25、0.5 mg/ml の濃度で検量線を描いた。 ・分子量測定(4 次構造予測) 分子量測定及び 4 次構造予測は、 Hiload 16/60 Superdex 200 pg カラムを用いたゲ ルろ過カラムクロマトグラフィーにより行った。分子量マーカーとして、MW-GF1000 Kit (SIGMA)から、Tyroglobulin (669 kDa)、Apoferritin (443 kDa)、β-Amylase (200 kDa)、Bovine Serum Albumin (66 kDa)、Carbonic Anhydrase (29 kDa)を用いた。 ① Hiload 16/60 Superdex 200 pg カラムを用いて 20 mM Tris-HCl pH 8.0 + 150 mM NaCl で平衡化を行った。 ② 精製·濃縮したサンプルを 20 mM Tris-HCl pH 8.0 + 150 mM NaCl を用いて適切な 濃度で希釈してカラムに吸着させ、その保持時間を測定した。 ③ 上記の分子量マーカーに関しても同様の操作で保持時間を測定した。 ・Tm-AFase の活性測定 ·活性測定用反応液: 100 mM Tris-HCl (pH 7.0)、0.5 mM pNP-α-L-arabinfuranoside ① 活性測定用反応液 385 μl をキュベットに分注し、温度が 60°C に保たれている分 光光度計内に入れ 10 分間プレインキュベートを行った。 ② 最適な濃度に設定した酵素液を 10 μl 加え、ピペッティングにより均一にし、 60°C で反応を開始した。 ③ 波長 405 nm における吸光度の変化を経時的に観察した。 1.4 Tm-AFase の結晶化条件の探索 ・目的 41 Tm-AFase の X 線構造解析を行うため、高分解能の回折を与える結晶を得ること が必要である。そのため、1.3 節で精製した Tm-AFase のサンプルを用いて結晶化ス クリーニングを行い、結晶化条件を探索した。 ・結晶化条件のスクリーニング PCT (Pre-Crystallization Test, Hampton)キットを用いて Tm-AFase の結晶化条件を探 索に適するタンパク質の濃度を見積もった結果、約 14.7 mg/ml が適切な濃度である ことが分かった。まずは、Sparse Matrix Screen 法による結晶化スクリーニングを行 って、その後塩濃度や PEG 濃度の組み合わせたキット、Sparse Matrix Screen の derivative などを用いて追加的にスクリーニングを行った。結晶化条件のスクリーニ ングは、インテリプレート(Art Robbins)を用いて、シッティングドロップ蒸気拡散法 により行った(図 1-11)。使用した結晶化キットを以下に示す。 ·JCSG + Suite (QIAGEN) ·Structure Screen 1 & 2、SaltRX (Hampton Research) ·Wizard 1 & 2、Precipitant Synergy (Emerald BioSystems) ① インテリプレートの各ウェルにスクリーニングキットの各溶液を 70 μl ずつ分出 し、リザーバー溶液とした。 ② インテリプレートの各ドロップにサンプルとリザーバー溶液を各々1 μl ずつ混ぜ た。 ③ Crystal Clear Sealing Tape (Hampton Research)を用いてインテリプレートを密閉し、 25°C に静置した。 ・結晶化条件の精密化 結晶が得られた条件について、さらに大きく質の良い結晶を得るため、沈殿剤濃 度、pH、タンパク質濃度、結晶化方法などについて精密化を行った。 1.5 X 線回折データ測定 ・目的 42 1.4 節で得られた結晶について、X 線を照射して回折イメージを測定した。放射線 による結晶の損傷を防ぐために、95 ~ 100 K の窒素ガスでフラッシュクーリングし た。また、低温での結晶の凍結を防ぐため、抗凍結剤である 30% 2-メチル 2,4-ペン タンジオール(MPD)、または 20%グリセロールを含む結晶化溶液に結晶を浸透させ た後、1.0 Å の波長でデータ測定を行った。 ① 3 Well Spot Plate (Hampton Research)のウェルに結晶化リザーバー溶液を 100 μl 入 れた。各ウェルの抗凍結剤の濃度を少しずつ上げ、終濃度を 20%にした。 ② 結晶化プレートから結晶を慎重にすくい、ウェルのリザーバー溶液に回収した。 準備した 0、10、20%のグリセロールを含む結晶化溶液にステップワイズ法で順 に移して結晶に抗凍結剤の濃度を上げた。 ③ 結晶をクライオループ(Hampton Research)にマウントし、100 K のクライオスト リームの中に設置して瞬間的に冷却し、X 線を照射して回折データイメージを 測定した。 X 線回折データ測定は、つくば市·高エネルギー加速研究機構(KEK)の放射光施設 Photon Factory (PF)実験ステーション BL5A、BL17A、NW12A、NE3A で行った。 1.6 分子置換法による初期構造決定及び構造精密化 ・目的 タンパク質の立体構造を決定する際に位相問題を解決することが必要である。X 線回折データは強度データのみであり、位相情報は失われてしまうからである。そ の位相を決定するために、本研究では分子置換法(Molecular Replacement、MR)を用 いた。分子置換法は、アミノ酸配列の同一性が高い他のタンパク質の立体構造をモ デルとして、計算により位相を決定する方法である。 ・分子置換による初期構造の決定 Geobacillus stearothermophiles 由来のアラビノフラノシダーゼ (Gs-AFase)は TmAFase と高いアミノ酸配配列の同一性を示すため、分子置換のモデル構造として用 43 いられると判断した[18]。Tm-AFase と Gs-AFase のアミノ酸配列同一性は約 35.1%で あり、他の AFase より最も類似の構造を持っていると予測される(表 1-3)。Gs-AFase の pdb フ ァ イ ル (1PZ2) と Tm-AFase の ア ミ ノ 酸 配 列 を 用 い て 、 SWISS-MODEL (http://swissmodel.expasy.org/)にてホモロジーモデリングを実施し、モデリング構造を 習得した。このモデリング構造を用いて、CCP4i の Molrep プログラムで分子置換を 行い[20]、Tm-AFase の初期構造を決定した。 ・構造精密化 分子置換により初期構造を決定したが、その構造が不完全であったため、 Arp/wArp[21]を用いてもう 1 回自動モデル構築を行った。その後、Refmac5 で自動 精密化行い、Coot を用いてペプチド鎖の手動構築及び水分子の処理など細かい精密 化を行った。この過程を繰り替えすことによって R 値が下がった最終構造を得るこ とができた。 ・構造解析 本研究で得られた Tm-AFase の全体構造及びドメインなど構造の新規性や有意性 を解析するために Dali Server (http://ekhidna.biocenter.helsinki.fi/dali_server/)を用いた。 また、Tm-AFase の立体構造に関するすべての図は PyMOL を用いて描いた[22]。そ の他に解析に用いたサーバやプログラムなどを以下に示す。 ·ExPASy Proteomics Server (http://www.expasy.ch/cgi-bin/protparam) ·Matthews Probability Calculator (http://ruppweb.dyndns.org/mattprob/) ·MOLPROBITY (http://molprobity.biochem.duke.edu/) ·Cremer-Pople parameter calculator (http://asfushi.hp.infoseek.co.jp/) ·PDBe PISA Server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) 1.7 基質複合体の構造解析 ・目的 基質認識部位の同定及び糖の加水分解及び重合反応メカニズムを明らかにするた め、本酵素の反応産物である L-アラビノース (L-Ara)との複合体の構造解析を行っ た。また、アラビノフラノシダーゼの本来の基質ではないが、Tm-AFase での活性が 44 あると報告されている pNP-Xyl の反応産物 D-キシロース(D-Xyl)との複合体の構造 解析も試みた[17]。 ・ソーキング法 1.4 節で得られた Tm-AFase の結晶についてソーキング法による複合体の構造解析 を試みた。用意した抗凍結剤を含むリザーバー溶液に終濃度 100 mM になるよう基 質 L-Ara を添加し、1.5 節と同様に結晶に浸透させた後 X 線回折データを測定した。 D-Xyl は Tm-AFase の本来のリガンドではないため、複合体構造を得るのが大変であ ったが、D-Xyl をほぼ飽和の濃度になるようにして、D-Xyl 自体が抗凍結剤になる ようなバッファーを利用した。 ・Xyl-S-DMT の共結晶化 本研究の目的の 1 つである Tm-AFase と Xyl-β-DMT の反応様式を明らかにするた めに、基質アナログである Xyl-S-DMT との共結晶を行った。タンパク溶液に 1 mM の Xyl-S-DMT を入れたものについて、 Structure Screen 1 & 2 、JCSG+ Suite、 Precipitant Synergy キットを用いて結晶化スクリーニングを行った。 1.8 Automated-docking 法による DMT 誘導体の触媒様式の予測 ・目的 Tm-AFase と DMT 誘導体による糖の合成反応(重合反応)のメカニズムを解明する ためには、DMT 誘導体との複合体構造が必要である。本研究では、アラビノース及 びキシロースからなる DMT 誘導体をモデリングで作成し、Tm-AFase の構造との結 合様式を Automated-docking 法により予測した。 ・Automated-docking Automated-docking のために、まずリガンドモデルとして DMT-Ara と DMT-Xyl を 設 計 し た 。 リ ガ ン ド モ デ ル の 構 築 は 、 PCModel プ ロ グ ラ ム (Serena Software, Bloomington, IN)を用いて行った。DMT-Ara のアラビノース環は、Gs-AFase 構造中 [18]の pNP-Ara の構造に基づいて 4E-conformation になるように設計した。また、 DMT-Xyl のキシロース環の配座異性体(conformer)は、GH 酵素のミカエリス複合体 45 構造で見られるコンフォメーション(B3O、14B、2SO、1S5 など)になるように複数のモ デルを作成したが、実際の結果ではそのうち 1S3-conformation を取っている DMT-Xyl を用いた結果が、最もよいクラスター値(Number of conformations in cluster)や低い自 由エネルギー値(Estimated free energy)を示したため、このコンフォメーションを採用 した。Automated docking は、AUTODOCK 4.0 プログラムを用いて行った[23]。TmAFase の構造から水分子やリガンドを全て削除した後、水素をつける作業により、 タンパク質構造の準備を行った。その後、グリットパラメータマップの作成を行っ たが、リガンドの探索範囲の xyz ボックスの大きさは 40×40×40 くらいになるように 作成した。256 回の Lamarckian genetic algorithm (LGA)に基づいた Automated docking を行って、その結果を RMSD < 2.0 Å でクラスタリングした。 46 結果と考察 1.9 組み換え Tm-AFase の精製 ・大量発現と精製 Tm-AFase の発現系は、宮崎らにより構築されており一定の条件で発現することも 確認されていた。本研究では、結晶化に向けた大量発現のため、宿主大腸菌株や培 養時間などを検討した結果(詳細は割愛する)、1.3 節に述べた条件で最も大量の TmAFase の発現が確認されることが分かった。Tm-AFase の大量発現、精製結果を表 14 に示す。熱処理、Ni カラムクロマトグラフィーによる精製を行ったが、その後、 熱処理の条件を少し調整し(80°C→70°C)、ゲルろ過クロマトグラフィーを加えて精 製を行った。その 3 ステップにおける SDS-PAGE を図 1-12 に示す。70°C 、30 分の 熱処理によって大腸菌由来のタンパク質をほとんど除去し、さらに Ni カラムクロマ トグラフィー及びゲルろ過クロマトグラフィーによって単一の目的産物を得ること ができた。精製の結果、3 L の培養から高純度に精製された約 10 mg の Tm-AFase が 得られた。 ・分子量測定/4 次構造予測 Tm-AFase の溶液中の分子量を測定し、その 4 次構造を予測した。標準タンパク質 分子量マーカーの溶出パターンを図 1-13(a)に、マーカーと Tm-AFase の溶出パター ンの関係を図 1-13(b)にプロットした。溶出から見積もられた Tm-AFase の分子量は 約 220 k であり、アミノ酸配列による Tm-AFase の分子量は約 55 k であったため、 Tm-AFase は溶液中で 4 量体を形成していると考えられる。この結果に関しては 1.12 節の「構造に関しての考察」で詳しく述べる。 1.10 Tm-AFase の結晶化条件の探索結果 精製した Tm-AFase に対して結晶化条件のスクリーニングを行った。PCT の結果 から結晶化に適したタンパク質の濃度を約 14.7 mg/ml に設定し、1.4 節に記述した ように JCSG + Suite (QIAGEN)、Structure Screen 1 & 2、SaltRX (HR)、Wizard 1 & 2、 Precipitant Synergy (EB)などのキットを用いてスクリーニングを行った。沢山の条件 47 で結晶が得られたが、X 線回折データ測定に適する綺麗な結晶は少なかった。その うち、外見的に綺麗で質の良かった結晶が作られた条件を以下の方に示す。 ·0.2 M Potassium nitrate, 20% PEG 3350 (図 1-14(a)) ·0.1 M Cacodylate pH 6.5, 0.2M MgCl2, 10% PEG 3000 (図 1-14(b)) 上記の条件で作成した結晶について X 線回折データ測定を行ったが、見た目は綺 麗な結晶であるものの、反射点の数が少なく構造解析に至るデータを得ることはで きなかった。そこで、違う結晶化条件を見つけるためにもう 1 回スクリーニングを 行った結果、異なる条件で新たな結晶を得ることができた。 ·0.1 M Sodium citrate, 0.1 M Tris-HCl pH 7.0, 0.2 M NaCl (図 1-14(c, d)) 上記の条件で得られた結晶は、再現性もよく、沢山の結晶を作成することが可能 であった。更なる良い質の結晶を作るために、Sodium citrate (0.05 mM ~ 1.15 mM)と NaCl (0.1 ~ 0.3 M)の濃度、Tris バッファーの pH (pH 7.0 ~ 8.0)などを変えながら結晶 化条件精密化を行い、複数の綺麗な結晶を取得した。これらの結晶は、シッティン グドロップ蒸気拡散法及びハンギングドロップ蒸気拡散法により、20°C で 1 ヶ月間 放置することで作られた。これらの結晶を用いて X 線回折データ測定を行った。 1.11 構造解析の結果 高エネルギー加速器研究機構(KEK)の放射光施設 Photon Factory (PF)実験ステーシ ョン AR-NW12A を利用し、得られた結晶に対して X 線回折データの測定を行った。 回折データについての解析及び処理は、HKL2000 を用いて行った。Tm-AFase の最 大分解能は 1.8 Å であり、空間群は P21 、Native 結晶の単位胞のパラメーターは a=101.515、b=160.647、c=155.735 (Å)、α=γ=90°、β=91.749°であった。VM=4.05 Å3/Da、 Vsolvent=69.6%であり、非対称単位中に 6 分子の Tm-AFase が入っていた。X 線回折デ ータの測定結果を表 1-5 に示した。 ・Tm-AFase の初期構造構築及び構造解析 1.6 節でも述べたように、Tm-AFase と最も高いアミノ酸配列同一性を持つ GsAFase との分子置換で立体構造の初期位相を決定した。Gs-AFase をモデル構造に Molrep を用いて分子置換を行ったところ、wRfac=0.621 となった。その後、Refmac5 によるマップとペプチドの自動精密化を行った結果が R/Rfree=44.5/48.2%であったが、 48 実際その構造を観察したところ、Tm-AFase は非対称単位中で 7 量体となっていた。 7 量体構造は非常に珍しいので、各単量体の間の相互作用や結晶のパッキングなど を調べたところ、7 つの分子中 1 分子が空間的な障害を起こしていることが分かっ た。1 つの分子を削除した 6 量体が本酵素の非対称単位中の四次構造だと判断して、 6 量 体 の 構 造 に 対 し て Refmac5 で の 自 動 精 密 化 を も う 1 回 行 っ た 結 果 、 R/Rfree=42.1/45.6%まで下がった。ARP/wARP でペプチド鎖を自動構築したところ、 主鎖 43 本で 2743/2746 残基がつながり、99%の構築率を示した。また、Refmac5 に よる精密化や Coot による手動構築を繰り返し、最終的に R/Rfree=17.9/19.9%の構造を 得ることに成功した。精密化の結果を表 1-5 に示した。 ・基質複合体の構造解析 Tm-AFase の反応産物である L-アラビノースと、本来の基質ではないが Tm-AFase に対しての微弱な活性を持つ pNP-Xyl の反応産物 D-キシロースとの複合体構造解析 のため、KEK-PF AR-NW12A にて波長 1.0000 Å で X 線回折データの測定を行った。 D-キシロースは本来のリガンドではないため、複合体構造を得るのが困難であった が、非常に高濃度のキシロースの使用と長時間ソーキングすることによって、その 複合体データを収集することができた。両複合体構造の最大分解能は各々2.15、2.3 Å であり、Native Tm-AFase と同じ空間群の P21 を示した。単位格子(Unit cell)の情報 などは Native Tm-AFase とほぼ一緒であった(表 1-5)。複合体構造解析は、Native TmAFase の構造を鋳型として Molrep を用いた分子置換により行った。基質結合部位で 見られた各リガンドの電子密度マップに従い、Coot での手動構築にてリガンドの構 造を明らかにした。詳しい基質の結合様式に関しては 1.12 節で論じる。Refmac5 に よる自動精密化や Coot による手動構築を繰り返して行った結果、最終的に R/Rfree が 16.9/20.6%、16.9/20.9%であった。複合体構造の X 線回折データ測定と精密化の結果 を表 1-5 に示した。 1.12 構造に関する考察 ・全体構造 1.9 節の Tm-AFase の 4 次構造予測結果から、Tm-AFase は溶液中で四量体を形成 していることが分かった。しかし、構造解析の結果からは、Tm-AFase は非対称単位 で六量体を形成していた(図 1-15(a))。これは酵素の形が原因となるゲルろ過クロマ トグラフィーの誤差から起因すると考えられる。ゲルろ過クロマトグラフィーは、 タンパク質が大きいほど早く溶出されるが、そこには大きさだけではなく形も重要 49 な要因であることが知られている。一般的に細長いタンパク質が丸いタンパク質よ り早く溶出することが分かっている。図 1-15(a) の Tm-AFase の全体構造を見るとそ の六量体が丸くなっており、hydrodynamic radius が小さくなることにより本来の溶 出時点より遅れて出てきたために分子量計算からは四量体と考えられる。実際に当 研 究 室 で は 同 じ 現 象 の 結 果 に つ い て 論 文 で 議 論 し た 履 歴 が あ る 。 Thermus thermophiles A4 由来の β-ガラクトシダーゼ(A4-β-Gal)はゲルろ過による分子量計算で は二量体と予想されたが、結晶構造解析の結果からは三量体を形成していたという ことが報告されている[24]。 Tm-AFase の六量体は、3 つの二量体で形成される「Trimer of dimer」であると考え られた。Chain A と Chain D で構成される二量体構造は、その 2 分子間に強い相互作 用をしていた(図 1-15(b))。PDBe PISA server を用いて 6 つの各単量体間の境界面に ついて調べてみたが、3 つの二量体間境界面(A-D, B-E, C-F)の平均面積は 616 Å2 であ るものの、6 つの三量体間境界面(A-B, B-C, C-A, D-E, E-F, F-D)は 639 Å2 を示した(図 1-16)。Tm-AFase で見られる六量体構造は、他の GH51 AFase でも類似的に見られた。 また、各単量体の表面には Tris 分子が 1 個ずつ総計 6 個の Tris 分子が構造の表面で 見られた(図 1-15(a))。この Tris 分子は、結晶化用のバッファーから由来するもので あり、各三量体の境界面に位置していたため、Tm-AFase の「Trimer of dimer」の構造 形成のパッキング相互作用に寄与していると考えられる。ちなみに、本実験で使わ れた酵素緩衝液は、Tris-HCl バッファーを使用したが、1.10 節の結果のように作ら れた結晶がすべて構造解析に使えるものではなかったということから、Tm-AFase の 高分解能が得られる結晶の結晶化には Tris が入っている条件が必須ではないかと考 えられる。また、リガンドフリーの Tm-AFase の構造では、51 個のエチレングリコ ール(Ethylene glycol, EG)が見られた。この EG は、抗凍結剤からのものであり、そ のほとんどがタンパク質の表面に結合していた。しかし、そのうち 18 個の EG 分子 は、活性中心に結合しているのが見られた。これに関しては、以降で詳しく論じる。 Tm-AFase の単量体は、N 末端の(β/α)8 TIM barrel ドメイン(Ser2-Ser357)と C 末端の β-sandwich ドメイン(Gly358-Leu484)の 2 つのドメインで構成されている(図 1-17)。 N 末端ドメインは GH の酵素でよく見られる典型的な TIM barrel 構造をとっており、 本酵素の触媒残基(Glu172、Glu281)がこのドメインに位置していた。C 末端ドメイン は、12 本の β-strand からなっており、直接的には酵素の機能に関与してないが、 Val367-Phe391 からなる β-hairpin motif が活性中心の周りを囲んでいた。この βhairpin motif は GH51 での保存性が無く、Thermotoga 属由来の AFase にのみ存在す るモチーフであった(図 1-4)。 ・活性中心及び複合体構造 50 N 末端の TIM barrel ドメインに位置している Tm-AFase の活性中心には、酸/塩基 触媒(acid/base catalyst)としての Glu172 と求核性残基(nucleophile)としての Glu281 が 存在しており、芳香族環をもついくつかの残基により疎水性ポケットを形成してい た。Native (リガンドフリー) Tm-AFase では、抗凍結剤由来の 3 つの EG 分子が活性 部位のサブサイト-1、+1、+2 に結合していた(図 1-18)。 アラビノース複合体構造では、3 つのアラビノース(α-L-arabinofuranose)がサブサ イト-1、+2、+3 に、結晶化バッファー由来の 1 つの Tris 分子がサブサイト+1 に結合 していた(図 1-19)。また、キシロース複合体構造では、1 つのキシロース(β-Dxylopyranose)がサブサイト+1、1 つの Tris 分子がサブサイト-1 に結合していた(図 120)。各基質の|Fobs| - |Fcalc|電子密度マップからは、糖のコンフォメーションとその位 置を決定することができた。Tm-AFase の活性中心に結合しているすべてのアラビノ ースとキシロース分子は、各々E3、4C1 の最も安定的な糖フラノース及びピラノース 環状配座を取っていた。糖ピラノース環の配座は、6 員環構成原子の 3 次元ユーク リッド空間座標をフーリエ変換し極座標系に射影することで、糖ピラノース環状配 座を定量的に表現する手法である Cremer-pople 法で表現できる(図 1-21)[25]。糖質関 連酵素の反応機構を解析する上で、基質である糖分子の動的拳動は重要である[26]。 各複合体構造での基質認識機構を詳しく見てみると、アラビノース複合体のサブ サイト-1 に結合しているアラビノース分子は、Glu26、Asn171、Glu172、Tyr237、 Glu281、Gln326 の側鎖及び Asn71 の主鎖と直接水素結合をしていた。これらは、 Gs-AFase のサブサイト-1 での結合様式と非常に似ていた(後で詳しく説明する)。サ ブサイト+2、+3 に結合しているアラビノース分子は、O1 ヒドロキシルが続くサブ サイト(+3 と+4)に向かってないため、不規則に(本来の結合とはことなる向きで)位 置しているように見られる。またアラビノース環の酸素原子(O4)がお互いに接して いることもこれらのアラビノース分子の結合が artifical なものである理由の 1 つで あると考えられる(図 1-19)。 一方、キシロース複合体では、キシロース分子がサブサイト+1 で Asn71、Glu172、 サブサイト-1 の Tris 分子、水分子と水素結合を形成しており、Trp177 の側鎖はキシ ロース環の疎水性の面とスタッキング相互作用をしていた。これらの結果から TmAFase の基質結合部位は主に Trp177 と Tyr237 によって疎水性のポケットを形成して いると見られる。 アラビノース分子がサブサイト-1、キシロース分子がサブサイト+1 に結合してい る各々のリガンドの結合様式から、これらの結合様式は天然に存在するアラビノキ シラン(arabinoxylan)の α-1,2-あるいは α-1,3-アラビノフラノシル(arabinofuranosyl)側 鎖に当たると考えられる。 しかし図 1-22(c, d)に示した Tm-AFase の分子表面図から、 基質結合ポケットが比較的に狭いため、アラビノキシランなどの巨大ポリマーを結 合しにくく、Tm-AFase はアラビノキシランそのものを加水分解できないと予測され 51 る。実際に、Tm-AFase は in vivo でアラビノキシランのポリマーを基質にできない ことが分かっている[9]。これらの結果から、Tm-AFase はシグナル配列を持たない 細胞内酵素であり、この酵素は細胞内のキシロオリゴ糖(xylooligosaccharide)のアラ ビノフラノシル側鎖を加水分解すると考えられる。 ・類似構造との比較 Tm-AFase の分子置換モデルとして用いた Gs-AFase は、GH51 AFase の中で最も研 究が進んでいる酵素であるため、両酵素の活性中心の比較を通して両酵素の違い、 Tm-AFase の特徴などを調べた。 PDBe-Fold server によると Tm-AFase は Gs-AFase (1PZ3、Q-score=0.71、 RMSD=1.45 Å for 458 residues)と非常に類似した構造をとっていた。図 1-22(a)には両 酵素の活性中心の重ね合わせを示した。Tm-AFase はサブサイト-1 にアラビノース 分子、サブサイト+1 にキシロース分子が、Gs-AFase は Ara-α1,3-Xyl が結合している 構造を用いた。サブサイト-1 でアラビノースと相互作用をしている周りのアミノ酸 残基は、両酵素でほとんど一致していた。また、両酵素のサブサイト+1 側のキシロ ース分子を比較してみると Gs-AFase より Tm-AFase の方が 60°くらい回転している ことが見られた。両酵素でのこのようなキシロースの結合様式から、Tm-AFase と Gs-AFase は異なる基質認識機構(特にサブサイト+1 側)をとっていると考えられる。 GH51 AFase で保存されているトリプトファン残基(Tm-AFase では Trp177、Gs-AFase では Trp180)は両酵素のサブサイト+1 で疎水的プラットフォームを形成していた。 図 1-22(b)は、Gs-AFase の pNP-Ara との複合体構造の分子表面図を表している。GsAFase と比べて、Tm-AFase は比較的狭い基質結合ポケットを形成していた(図 122(c))。それは、Tm-AFase が持つ二つの芳香族環(Phe378、Tyr174)と Asp213 と Arg256 の間のイオン結合によると考えられる。Phe378 は Tm-AFase の β-hairpin motif 挿入ループ上に位置していたため、この基質結合ポケットは Tm-AFase に特異 的であると考えられる。このように Tm-AFase の基質結合ポケットは、その形と性 質から硬貨スロットのような疎水性ポケットとして「Coin slot like hydrophobic cavity」 と名づけた。 ・Automated-docking による DMT 誘導体の結合 本研究では Tm-AFase の新規構造の解明以外にも、DMT 誘導体の複合体構造の解 明を目指した。東北大学から提供していただいた基質アナログ DMT-S-Xyl は、溶解 度が低いために、ソーキングや共結晶化に必要な高濃度の溶液を作ることができず、 その複合体構造は得ることができなかった。そこで、本研究では DMT-Xyl、Ara52 α1,3-Xyl、Ara-α1,2-Xyl と Tm-AFase の結合様式を推測するために Automated docking 解析を行った。Tm-AFase に対して見つかった各基質の最も信頼できるコンフォメー ションで、一番上位にランクされた結果を表 1-6 に示した。図 1-23(a, b)には予測さ れる Ara-α1,3-Xyl、Ara-α1,2-Xyl の Tm-AFase への結合様式を示したが、その結合様 式は Gs-AFase の Ara-α1,3-Xyl との複合体構造と非常に似ていた(図 1-22(a))。Araα1,3-Xyl と Ara-α1,2-Xyl ドッキングモデル中、キシロース分子の結合様式を比べた ところ、後者のキシロース分子が約 60°くらい回転しており、糖のリングと同じ面 から Trp177 とスタッキング相互作用を形成していた。これらの結果から、図 1-20 で示したキシロース複合体構造でのサブサイト+1 に結合したキシロースの結合様式 は Ara-α1,3-Xyl、Ara-α1,2-Xyl の中間体の結合様式を取っていると予測される。 DMT-Xyl と Tm-AFase の推測される結合様式を図 1-23(c)に示した。Tm-AFase の サブサイト-1 側を占めているキシロピラノース部分は、Gs-AFase での結合様式とほ とんど一致していた。サブサイト+1 における結合様式を説明するために図 1-23(d)に Gs-AFase の pNP-アラビノースとの複合体構造を示した。DMT-Xyl の DMT 基は 4 と 6 の位置に 2 つのメトキシ基を持っているために、pNP 基より大きくなっている。 しかし、ドッキングの結果から見られる DMT 基の結合様式では、Trp177 の芳香族 インドール側で形成された疎水性のプラットフォームに強く結合していることが見 られた。これらの結果から、Tm-AFase の「Coin slot like hydrophobic cavity」は、DMT 糖をドナーとアクセプター両方として受容できるような形状をとっていると考えら れる。 ・Tm-AFase の熱安定性機構 本章の序論でも述べたように、Tm-AFase は現在まで知られているアラビノフラノ シダーゼの中で最も耐熱性かつ好熱性の酵素である。ここでは、本研究で明らかに した Tm-AFase の立体構造に基づき、Tm-AFase を構造生物学的な観点から見た熱安 定性機構について調べた。一般的に知られている好熱菌タンパク質の耐熱化機構に は、高温で不安定なアミノ酸の除去、α-へリックスの安定化、疎水性相互作用の最 適化、立体構造のコンパクト化、 イオン結合,イオン結合ネットワークによる静電 相互作用の強化、サブユニット間相互作用の強化、不完全な熱変性状態などが上げ られる[27]。 構造既知の GH51 AFases の至適温度を図 1-24 に示した。好熱菌(thermophile)であ る Clostridium、Thermobacillus、Geobacillus 由来のアラビノフラノシダーゼ CtAFase[28]、Tx-AFase[29]、Gs-AFase[18]の至適温度は各々82°C、75°C、70°C であり、 中温菌(mesophile)に属する Bifidobacterium 由来の Bl-AFase は、論文報告が無いため 正確な至適温度を調べることはできなかったが、20 ~ 45°C の付近であると予測され 53 る。これらの中で好熱菌由来の Gs-AFase と中温菌由来の Bl-AFase との熱安定性機 構を比較してみた。表 1-7 に 3 つの酵素のアミノ酸組成を示した。中性の極性アミ ノ酸のうち Gln、Asn は高温環境化で脱アミド化を受けやすくこの脱アミノ化反応 は、同じタンパク質中の Thr、Ser によって促進される[30]。Tm-AFase の極性アミノ 酸の割合は 16%で Gs-AFase と Bl-AFase の 16.6%、20.7%より低くなっていることが 分かった。また、分子内部の疎水コーアを形成するアミノ酸(Ile、Leu、Val)はその 疎水コアのパッキング改善によって分子内部の疎水性相互作用を増大して耐熱性を 上昇することが知られている[31]。それらのアミノ酸残基の割合は Tm-AFase が 22.9%であるため、22.4%と 16.5%の含量をもつ Gs-AFase と Bl-AFase より高いこと から疎水性残基の相互作用が最適化されており、Tm-AFase の耐熱性に寄与している と考えられる。 また、Tm-AFase では図 1-25 に示したようにその耐熱性機構を獲得するために重 要ないくつかの特徴を持っていた。一般的に、好熱菌タンパク質は、常温菌由来の タンパク質より小さい傾向がある。これは立体構造のコンパクト化が耐熱性機構に 影響を及ぼしていると考えられるが、Tm-AFase の場合はいくつかのループが GsAFase と Bl-AFase より短縮化されていた。それ以外にも、分子内イオン結合の数が 28 個など他の酵素と比べて耐熱化によく見られる特徴を持っていた。さらに、TmAFase のサブユニット間の相互作用の強化(Chain A と Chain D など二量体構造間の 強い相互作用)、Thermotoga 属だけに見られる β-hairpin motif 構造が活性中心構造を 囲んでいることなどが構造の熱安定性機構に強く関与していると考えられる。ただ し、ループ上の Pro 残基の数は、14 個と他の酵素と比べて少なかった。 これらの沢山の構造上の理由で Tm-AFase は、今まで知られているアラビノフラ ノシダーゼの中で最も高い耐熱性を持っていると考えらる。したがって、このよう な耐熱性酵素を用いた研究及び産業上の利用が期待される。 1.13 本章のまとめ 本研究では、Thermotoga maritima MSB8 由来のアラビノフラノシダーゼ、TmAFase の立体構造を明らかにした。Tm-AFase は合成基質である DMT 誘導体(DMTXyl)と反応して糖の加水分解反応だけではなく、糖重合反応を触媒することによっ て新規のオリゴ糖を生成することが報告されている。Tm-AFase の構造は、構造既知 の類似酵素である Geobacillus stearothermophiles 由来のアラビノフラノシダーゼ、 Gs-AFase (アミノ酸配列同一性: 35.1%)の構造をテンプレートモデル構造にし、分子 置換法により初期構造を明らかにした。Native Tm-AFase の構造、アラビノース及び キシロースとの複合体構造の構造解析を通して Tm-AFase が持つ活性部位の特徴、 54 基質結合様式などを明らかにした。Tm-AFase の基質認識部位のサブサイト-1 は、 他の GH51 アラビノフラノシダーゼとほとんど同じであったが、そのファミリーで 高い保存性を持つ Trp177 からなるサブサイト+1 側の疎水性フラットフォームは Tm-AFase で特徴的に形成されていた。Tm-AFase は他の AFase のサブサイト+1 より 非常に狭い基質結合ポケットを形成していたが、その疎水性の基質ポケットは TmAFase が持つ 2 つの疎水性残基(Phe378、Tyr174)と Asp213-Arg256 からなるイオン結 合により形成されている。特に 378 番目のフェニルアラニン残基は、Thermotoga 属 のみで見られる特徴的な β-hairpin motif の中に存在していることから、この β-hairpin motif 挿入ループは Tm-AFase の独特な基質結合部位を形成する重要な役割をしてい ると考えられる。また、この β-hairpin motif は活性中心の周りを囲んでいる形をし ているため、この酵素の熱安定化機構にも寄与していると予測される。 一方、DMT 誘導体との複合体構造解析を通して、Tm-AFase と DMT 誘導体の間 で行われる糖重合反応の構造生物学的解析を行うことを試みたが、技術的な問題か ら複合体構造を得ることはできなかった。そこで、本研究で明らかにした Tm-AFase の立体構造に基づいて、Automated docking 解析を行い、DMT-Xyl との結合様式を予 測した。その結果、2 つのメトキシ基によりかさ高くなっている DMT 部分が、TmAFase の狭い疎水性プラットフォームの基質ポケットに強く結合していることが推 定された。他の AFase より狭い基質結合ポケットを持っているにもかかわらず、 Tm-AFase のサブサイト+1 は DMT 糖を受容するために最適化されていると考えられ る。 Tm-AFase は、最も高い耐熱性を持つアラビノフラノシダーゼであるが、本研究の 構造解析よりその耐熱性機構が明らかになり、これから DMT 糖を用いたオリゴ糖 合成にも利用されることが期待する。 55 図表 a) 乾重量(%) セルロース ヘミセルロース リグニン 40-60 20-40 10-25 b) 植物 六炭糖 五炭糖 リグニン 灰分 広葉樹 針葉樹 農業廃棄物 39-50% 41-57% 30-42% 18-28% 8-12% 12-39% 15-28% 23-27% 11-29% 0.3-1.0% 0.1-0.4% 2-18% 表 1-1 植物細胞壁の構成成分 a) 植物細胞壁におけるセルロース、ヘミセルロース、リグニンの割合 b) 植物の細胞壁成分の割合: 植物によってその細胞壁の構成成分は異なる。ヘミセ ルロースの主な成分は、広葉樹ではアラビノキシラン(五炭糖)、針葉樹ではグルコ マンナン(六炭糖)であるので、広葉樹の方が五炭糖の割合が多くなる。 56 GH family Enzyme Mechanism Clan Structure (AFase) retaining none inverting GH-F Streptomyces avermitilis Selenomonas ruminantium Bacillus subtilis retaining GH-A C. thermocellum G. stearothermophilus T. xylanilyticus, B. longum T. maritima, T. petrophila retaining none Aspergillus kawachii Not known retaining retaining GH-F none none Aspergillus niger β-glucosidase xylan 1,4-β-xylosidase β-N-acetylhexosaminidase GH3 glucan 1,3-β-glucosidase glucan 1,4-β-glucosidase exo-1,3-1,4-glucanase GH43 α-L-arabinofuranosidase β-xylosidase β-1,3-xylosidase α-L-arabinofuranosidase arabinanase xylanase galactan 1,3-β-galactosidase α-L-arabinofuranosidase GH51 endoglucanase GH54 GH62 GH121 GH127 α-L-arabinofuranosidase β-xylosidase α-L-arabinofuranosidase β-L-arabinofuranosidase β-L-arabinofuranosidase 表 1-2 アラビノフラノシダーゼの分類 57 Source organism Protein Identity (%) PDB ID Geobacillus stearothermophilus Gs-AFase 35.1 (182/519) 1PZ2, 1PZ3, 1QW8, 1QW9 [18] Clostridium thermocellum Ct-AFase 31.4 (173/551) 2C7F, 2C8N [28] Thermobacillus xylanilyticus Tx-AFase 32.3 (167/517) 2VRK, 2VRQ [29] Bifidobacterium longum Bl-AFase 28.8 (173/601) 2Y2W 表 1-3 GH51 AFase の種類と Tm-AFase との配列同一性 GH51 AFase の中で最も配列同一性が高い Gs-AFase を用いて分子置換による初期構 造を決定した。 protein (mg) activity (U) specific activity (U/mg) yield (%) 100 菌破砕液 786 409 0.52 熱処理 121 400 3.28 HisTrap HP 12.3 162 13.2 97.7 39.7 表 1-4 Tm-AFase の大量発現(3 L)と精製結果 58 Purification fold 1 6.31 25.4 Data Collection and Refinement Statistics Data set Ligand-free Ara complex Data collection PDB ID 3UG3 3UG4 Space group P21 P21 Unit cell parameters a (Å) 101.5 100.5 b (Å) 160.6 160.6 c (Å) 155.7 154.8 β (°) 91.8 92.4 50.00-1.80 50.00-2.15 Resolution (Å)a (1.86-1.80) (2.23-2.15) Total reflections 3,278,651 2,040,914 Unique reflections 446,597 266,042 Completeness (%)a 97.4 (95.6) 100.0 (100.0) Redundancya 7.3 (6.6) 7.7 (7.6) Mean I/σ (I)a 34.1 (3.4) 20.8 (3.0) Rsym (%)a 6.1 (49.5) 10.8 (63.1) Refinement Resolution (Å) 30.14-1.80 29.99-2.15 No. of reflections 422,884 252,016 R-factor / Rfree (%) 17.6 / 19.6 16.9 / 20.6 No. of atoms 25,946 25,514 1,851 (Water), 2,436 (Water), 11 (Tris), No. of solvents 6 (Tris), 51 (EG), 1 (Na+) 31 (arabinose) RMSD from ideal values Bond lengths (Å) 0.013 0.020 Bond angles (°) 1.300 1.836 Ramachandran Plot (%) Favored 94.8 94.9 Allowed 4.6 4.2 Outlier 0.66 0.97 Data for the highest resolution shell are given in parentheses. Xyl complex 3UG5 P21 102.2 160.9 156.2 91.8 50.00-2.30 (2.38-2.30) 816,725 221,594 99.7 (98.7) 3.7 (3.4) 17.6 (2.5) 7.8 (38.0) 29.59-2.30 209,764 16.9 / 20.9 24,760 1,257 (Water), 12 (Tris), 15 (xylose) 表 1-5 X 線回折データ測定と構造精密化の統計値 59 0.021 1.738 94.9 4.1 0.97 Enzyme Ligand Number of conformations in cluster (rank #1) Estimated Free Energy (kcal/mol) Ara-α1,3-Xyl 89 % (227 / 256) - 7.13 Ara-α1,2-Xyl 75 % (192 / 256) -6.56 Ara-pNP 100% (256 / 256) -5.80 Ara-DMT 91% (233 / 256) -6.45 97% (249 / 256) -5.67 98% (252 / 256) -6.51 Tm-AFase 1 Xyl( S3)-pNP 1 Xyl( S3)-DMT 表 1-6 Automated docking 解析の結果 Tm-AFase Gs-AFase Bl-AFase Asn, Gln (deamidation) 6.7% 7.4% 7.1% Arg, Glu, Lys (charged residue) 20.2% 18.8% 14% Ile, Leu, Val (hydrophobic residue) 22.9% 22.4% 16.5% Phe, Trp, Tyr (aromatic hydrophobic resudue) 11.4% 10% 8.4% 表 1-7 アミノ酸合量による耐熱性機構の解析 60 「Kolpak,, F.J. et al., Macromolecu M ules, 1976. 9((2), 273-8」よ より抜粋 図 1-1 植物細胞壁 植 壁のモデルとセルロー ース繊維 a) 植物 物細胞壁のモデル: 基本骨格に相 基 相当するセ セルロース繊維に対し してその間 間にヘミ セルロ ースやリグ グニン(この の図では略 略)が存在す する構造をとっている る。さらに に、セル ロースやヘミセル ルロースは はペクチンに によって架 架橋される。 ルロース繊維 維の構造: グルコース スが規則的 的に並んでい いる。 b) セル c) グル ルコース間の水素結合 合: セルロー ースを構成 成しているグルコース スは、分子 子間およ び分子 子内に水素結 結合を形成 成しており、 セルロー ース繊維は強 強固なもの のになる。 61 図 1-2 細胞 成分の構造 胞壁構成成 a) セル ルロース b) 様々 々なヘミセル ルロース c) ぺク クチン 62 図 1-3 キシラ ランの構造とその分解酵素 キシラン主鎖を を黒で、側鎖のアラビノースを を緑で、糖以外 外の側鎖(アセチ チル基、フェルラ酸)を青で示した。 63 図 1-4 GH51 AFase の のアミノ酸 酸配列のアラ ライメント 64 「Miyazaki,, K., et al., Extremophile E s, 2005. 9(5)), 399-406」よ より抜粋 図 1-5 Tm-AF Fase の好熱 熱性及び耐熱 熱性 a) Tm--AFase の温 温度依存的 的活性測定。 。0.1 M Tris-HCl T (p pH 7.0)で、 1 mM pN NP α-Larabinoofuranoside を基質とし して測定を を行った。T Tm-AFase の至適温度 の 度は 90°C であり、 で 100°C で 20 分間反 反応するこ ことで 50% の活性が失 失活した。 b) Tm--AFase の熱 熱不活性化 化半減期測定 定。酵素液 液を各々90°°C (黒丸)、 100°C(白丸)でイ ンキュベートし、一定の時 時間ごとに活 活性を測定 定した。 65 図 1-6 Tm-AF Fase とキシ シロース誘 誘導体の酵素 素反応(TLC C)* Tm-AFase は DMT T-Xyl との反応により りいくつか かの DMT 糖を生成した 糖 た。しかし し、 pNP-Xyyl との反応 応では、生成 成物が見ら られなかった。 図 1-7 Tm-AF Fase と DM MT-Xyl の反 反応産物の HPLC 分離 離* Tm-AFase と DM MT-Xyl との の反応によ って生成さ された生成物に対して て HPLC を用いた を 分離を行った。 66 (* *図 1-5、1-66、1-7 のデータは東北大学正田研 研による実験 験データ) 図 1-8 NMR によ よるキシロオ オリゴ糖の の同定* HPLC に により分離 離された反応 応生成物の の構造を NM MR 解析を を用いて同定 定した。 67 図 1-9 DMT-β-Xyl D と Tm-AFaase の多糖合 合成反応 a) 合成 成基質 DMT T-β-Xyl と Tm-AFase T の多糖合成 成反応 b) 多糖 糖合成反応の生成物。Xyl-β(1→ →3)-Xyl-DM MT が 11%、Xyl-β(1→ →2)-Xyl-DM MT が 7%、X Xyl-β(1→3)--Xyl(1→2)--DMT また たは Xyl-β(1 1→2)-Xyl(1 1→3)-DMT T が 3%生成 成され る 68 図 1-1 10 Tm-AFaase 発現ベク クターpET2 28a(+) 業技術総合 合研究所の宮 宮崎博士に に提供してい いただいた た。 本ベクターは産業 69 図 1-11 結 結晶化方法 法の概念図 70 図 1-12 Tm-AFaase の大量発 発現·精製の の結果 処理、Ni カラムクロマ カ マトグラフ フィー後のサンプルの の純度を SSDS-PAGE で確認 a) 熱処 した。(ゲル濃度:: 7%、M: 分子量マー ーカー、C: 菌体破砕上清、H: 熱処理、W W: Ni カ ラフィー非 非吸着画分、 Elute: Ni--カラムクロ ロマトグラ フィー吸着 着画分) ラムクロマトグラ ク ラフィー時 時の UV モニ ニターリン ング結果、目 目的のタン ンパク質 b) Gel-ffiltration クロマトグラ が単一ピークとし して検出された。 71 図 1-13 ゲルろ過 過による Tm m-AFase の 4 次構造予測·分子量 量測定 準タンパク ク質分子量 量マーカー の溶出パタ ターン。分 分子量マー ーカーとし しては、 a) 標準 Tyrogloobulin 、 appoferritin 、 β-amylaaase 、 alcoh hol dehydrrogenase 、 BSA 、 carbonic c anhydraase の 6 つの の標準タン ンパク質を用 用いた。 b) マー ーカーと Tm-AFase T の溶出パタ の ターンのプロット化に により、Tm m-AFase の分子量 の を予測した。赤四 四角は Tm-AFase、青 青四角は標準 準タンパク質。 72 図 1-144 Tm-AFasee の結晶 a) 0.2 M Potassium m nitrate, 20 0% PEG 33550 b) 0.1 M Cacodylate pH 6.5,, 0.2M MgC Cl2, 10% PEG P 3000 の条件で作 の 作成した Tm m-AFase の結晶。両方とも も構造解析 析に至る X 線 線回折デー ータを得ることができ きなかった。 。 c) 実際 際の構造解析 析に用いら られた結晶は は、0.1 M Sodium cittrate, 0.1 M Tris-HCl pH 7.0, 0.2 M N NaCl の条件 件で作られ れた。 d) 複合 合体構造のデ データ測定 定に用いた結 結晶。c)と と同じ条件で で作られた た。 73 図 1-15 Tm m-AFase の全体構造 の a) Tm-AFase は、結晶格子 子中六量体(T Trimer of dimer)で存 存在していた た。黄色は は抗凍結 剤とし て用いたエ エチレング グリコール((EG)分子、紫色は結晶化バッフ ファー由来 来のトリ 子であり、三 三量体構造 造を強くする るパッキン ングの役割を をしている る。 ス分子 AFase の二 二量体(Chain n A と Chaain D)構造の の間には、強い相互作 作用が形成 成してい b) Tm-A た。 74 図 1-16 PDBe PISA A server によ よる Tm-AF Fase のイン ンターフェー ース解析 75 図 1-17 Tm m-AFase の の単量体構造 造とそのト トポロジ図 Tm-AFase の単量 量体は 25 本鎖の 本 β-strrand と 14 本の α-heliix からなり り、 GH 構造で典 構 型的に見られる TIM T barrel 個メインと と 12 本の β-strand からなる か β-ssandwich ドメイン ド の 2 つのドメイン ンで構成されている。 76 図 1-18 Tm m-AFase の活性中心 の AFase の活 活性中心には は、抗凍結 結剤由来のエ エチレング グリコール(EG) Ligand free Tm-A 分子が がリガンド として結合 合していた た。GH51 AFase で保 保存されて ている活性 性残基が Glu1722、Glu281 として存在 在しており 、Thermottoga 属 AF Fase が特徴 徴的持つ β-hairpin β motif が が保存され れており、活 活性中心の のサブサイト+3 の一部 部を形成し していた。E EG 分子 を緑、活性部位を を形成する残基を黄色 色、β-hairpin motif をシアンで表 表示した。 77 図 1-19 アラ ラビノース ス複合体構造 造 a) 反応 応生成物であ あるアラビ ビノース分子 子がリガン ンドとして Tm-AFasee の活性中 中心に結 合している。アラ ラビノース分子は、各 各々サブサ サイト-1、+ +2、+3 に結 結合し、結 結晶化バ ッファー由来のト トリス分子 子がサブサイ イト+1 に結 結合してい いた。黄色は はアラビノ ノース、 紫色はトリス分子 子を示す。 b) アラ ラビノースのオミット トマップを示 示した。 78 図 1-20 キ キシロース複 複合体構造 造 シロースと Tm-AFasee の複合体 体構造を表し した。キシ シロース分子 子はサブサ サイト+1 a) キシ に、トリス分子は はサブサイト-1 に結 結合していた た。キシロース分子を をシアンで で、トリ スを紫 紫色で示した た。 b) リガ ガンドであるキシロー ースのオミ ットマップ プを通して て、糖のコン ンフォメー ーション とその位置を決定 定することができた。 79 「F Fushinobu, S.., et al., Carb bohydr Res, 2008. 2 343(6)), 1023-33」よ より抜粋 図 1-2 1 Cremer-P Pople 法 a) IUPA AC 命名法に における代 代表的な配座 座の例 b) 糖ピ ピラノース環 環配座を球 球座標に転 換した図 (左)。左図 図の球を「北 北極」から投 投影した 図。θ と φ で表さ され、メルカ カトル図法 法のような表 表現もなさ される。 c) GH ファミリー ー別に、加 加水分解反応 応の過程で で、基質で である糖がど どのような なコンフ ォメーション変化 化をするか かが研究され れており、その変化を を b) (右)の の図の投影 影図上で 表した図。 80 図 1-22 Tm-AF AFase と Gs-AFase の比 比較 a) Tm-A AFase と Gs-AFase G の活性中心の の の重ね合わ わせ。Tm-A AFase はキシ シロース複 複合体構 造(シア アン: キシ ロース、紫 紫色: トリ ス)を、Gss-AFase は Ara-Xyl 複 複合体(オレンジ: Ara-α1,,3-Xyl)構造 造を用いた。 。 b) Gs-A AFase の分子 子表面図 c, d) Tm m-AFase の分子表面図 の 図。各々ア アラビノース ス複合体とキシロース ス複合体構 構造を示 した。 81 図 1-23 Autom mated dockinng によるリ リガンド結 結合様式の予 予測 予測される Tm-A AFase と各 各リガンドと との結合様 様式。a) Arra-α1,3-Xyll、b) Ara-α α1,2-Xyl、 c) DMT T-Xyl、d) Gs-AFase G の pNP-Ara 複合体構造 造 82 図 1-24 GH H51 アラビ ビノフラノシダーゼの の至適温度 Tm-AFase は現在 在まで知られ れている最 最も高温性の のアラビノフラノシダ ダーゼであ ある。 83 図 1-25 Tm-AFasee の構造生物 物学的耐熱 熱性機構 ループ プ(緑): Tm-A AFase、ルー ープ(グレイ イ): Gs-AFase Tm-AFase の Salt bridge 残基 基(黄)、Proo 残基(マゼ ゼンタ) 84 引用文献 1. Kolpak FJ & Blackwell J (1976) Determination of the structure of cellulose II. Macromolecules 9, 273-278. 2. McNeil M, Darvill AG, Fry SC & Albersheim P (1984) Structure and function of the primary cell walls of plants. Annu Rev Biochem 53, 625-663 3. Ward OP & Mooyoung M (1989) Enzymatic Degradation of Cell-Wall and Relat ed Plant Polysaccharides. Crit Rev Biotechnol 8, 237-274. 4. Coughlan MP, and Geoffrey P Hazlewood. (1993) Hemicellulose and hemicellula ses. Portland Press, London. 5. Gilbert HJ & Hazlewood GP (1993) Bacterial Cellulases and Xylanases. J Gen Microbiol 139, 187-194. 6. Koseki T (2006) ヘミセルロース分解酵素におけるアクセサリー酵素とその意 義. In 日本農芸化学会北海道支部・東北支部合同若手シンポジウム, 洞爺湖. 7. Saha BC (2000) Alpha-L-arabinofuranosidases: biochemistry, molecular biology a nd application in biotechnology. Biotechnol Adv 18, 403-423. 8. Miyanaga A, Koseki T, Matsuzawa H, Wakagi T, Shoun H & Fushinobu S (200 4) Crystal structure of a family 54 alpha-L-arabinofuranosidase reveals a novel c arbohydrate-binding module that can bind arabinose. J Biol Chem 279, 44907-44 914. 9. Huber R, Langworthy TA, Konig H, Thomm M, Woese CR, Sleytr UB & Stette r KO (1986) Thermotoga-Maritima Sp-Nov Represents a New Genus of Unique Extremely Thermophilic Eubacteria Growing up to 90-Degrees-C. Arch Microbiol 144, 324-333. 10. Achenbach-Richter L, Gupta R, Stetter KO & Woese CR (1987) Were the origin al eubacteria thermophiles? Syst Appl Microbiol 9, 34-39. 11. Manca MC, Nicolaus B, Lanzotti V, Trincone A, Gambacorta A, Peter-Katalinic J, Egge H, Huber R & Stetter KO (1992) Glycolipids from Thermotoga maritim a, a hyperthermophilic microorganism belonging to Bacteria domain. Biochim Bio phys Acta 1124, 249-252. 12. Bronnenmeier K, Kern A, Liebl W & Staudenbauer WL (1995) Purification of Thermotoga maritima enzymes for the degradation of cellulosic materials. Appl E nviron Microbiol 61, 1399-1407. 13. Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey E K, Peterson JD, Nelson WC, Ketchum KA, et al. (1999) Evidence for lateral ge ne transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399. 85 14. Meissner K, Wassenberg D & Liebl W (2000) The thermostabilizing domain of the modular xylanase XynA of Thermotoga maritima represents a novel type of binding domain with affinity for soluble xylan and mixed-linkage beta-1,3/beta-1, 4-glucan. Mol Microbiol 36, 898-912. 15. Xue Y & Shao W (2004) Expression and characterization of a thermostable beta -xylosidase from the hyperthermophile, Thermotoga maritima. Biotechnol Lett 26, 1511-1515. 16. Suresh C, Kitaoka M & Hayashi K (2003) A thermostable non-xylanolytic alpha -glucuronidase of Thermotoga maritima MSB8. Biosci Biotechnol Biochem 67, 23 59-2364. 17. Miyazaki K (2005) Hyperthermophilic alpha-L: -arabinofuranosidase from Thermo toga maritima MSB8: molecular cloning, gene expression, and characterization of the recombinant protein. Extremophiles 9, 399-406. 18. Hovel K, Shallom D, Niefind K, Belakhov V, Shoham G, Baasov T, Shoham Y & Schomburg D (2003) Crystal structure and snapshots along the reaction path way of a family 51 alpha-L-arabinofuranosidase. Embo J 22, 4922-4932. 19. Tanaka T, Noguchi M, Kobayashi A & Shoda S (2008) A novel glycosyl donor for chemo-enzymatic oligosaccharide synthesis: 4,6-dimethoxy-1,3,5-triazin-2-yl g lycoside. Chem Commun (Camb), 2016-2018. 20. Murshudov GN, Vagin AA & Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallo gr 53, 240-255. 21. Greaves RB, Vagin AA & Dodson EJ (1999) Automated production of small-mo lecule dictionaries for use in crystallographic refinements. Acta Crystallogr D Bi ol Crystallogr 55, 1335-1339. 22. DeLano WL (2004) Use of PYMOL as a communications tool for molecular sci ence. Abstr Pap Am Chem S 228, U313-U314. 23. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK & Olson AJ (1998) Automated docking using a Lamarckian genetic algorithm and an em pirical binding free energy function. J Comput Chem 19, 1639-1662. 24. Hidaka M, Fushinobu S, Ohtsu N, Motoshima H, Matsuzawa H, Shoun H & W akagi T (2002) Trimeric crystal structure of the glycoside hydrolase family 42 b eta-galactosidase from Thermus thermophilus A4 and the structure of its complex with galactose. J Mol Biol 322, 79-91. 25. Cremer D & Pople JA (1975) General Definition of Ring Puckering Coordinates. J Am Chem Soc 97, 1354-1358. 26. Fushinobu S, Mertz B, Hill AD, Hidaka M, Kitaoka M & Reilly PJ (2008) Co mputational analyses of the conformational itinerary along the reaction pathway of GH94 cellobiose phosphorylase. Carbohyd Res 343, 1023-1033 27. 赤沼哲史 (2009) 好熱菌のタンパク質はなぜ熱に強いか. 生化学 81, 1064-1071. 86 28. Taylor EJ, Smith NL, Turkenburg JP, D'Souza S, Gilbert HJ & Davies GJ (200 6) Structural insight into the ligand specificity of a thermostable family 51 arabi nofuranosidase, Araf51, from Clostridium thermocellum. Biochem J 395, 31-37. 29. Paes G, Skov LK, O'Donohue MJ, Remond C, Kastrup JS, Gajhede M & Mirza O (2008) The structure of the complex between a branched pentasaccharide and Thermobacillus xylanilyticus GH-51 arabinofuranosidase reveals xylan-binding de terminants and induced fit. Biochemistry 47, 7441-7451. 30. Wright HT (1991) Nonenzymatic deamidation of asparaginyl and glutaminyl resid ues in proteins. Crit Rev Biochem Mol Biol 26, 1-52. 31. Dong H, Mukaiyama A, Tadokoro T, Koga Y, Takano K & Kanaya S (2008) H ydrophobic effect on the stability and folding of a hyperthermophilic protein. J Mol Biol 378, 264-272. 87 第2章 新規二機能性フラビン酵素 Pseudomonas sp. AIU 813 由来 L-アミノ酸オキシ ダーゼ/オキシゲナーゼの二機能性反応機構に関する構造生物学的研究 (Structure-based study on catalytic mechanism of a novel bifunctional flavoprotein L-amino acid oxidase/monooxygenase from Pseudomonas sp. AIU 813) 88 序 2.1 Pseudomonase sp. AIU 813 由来の L-アミノ酸オキシダーゼ/オキシゲナーゼ ・L-リジン(L-Lysine) L-アミノ酸は、主に蛋白質やペプチドの構成成分として知られているが、それ以 外にも食品栄養分、医薬品の材料、代謝系の中間物質など様々な分野で使われてい る。そのうち、L-リジンは、人の正常的な成長のために必要な 8 個の必須アミノ酸 の 1 種類であり、人体内で合成されないため、食品としての摂取が必要である[1]。 体内のリジン代謝異常としては、高リジン血症(hyperlysinemia)が上げられる。高リ ジン血症は、リジン代謝に対する先天性代謝異常症(inborn error)であり、血中及び尿 中にリジンの著明な増量が見られ、リジン経口負荷により血中リジンの高値、復帰 遅延が見られることからリジン分解経路に障害が予想される症状である[2, 3]。この ように、体内におけるリジンの代謝及びその定量は、食品·医薬品分野で非常に興味 深いところである。 ・L-リジンオキシダーゼ(L-Lysine oxidase) L-リジンオキシダーゼは、L-アミノ酸酸化酵素(L-amino acid oxidase, LAAO)の代表 的な酵素の 1 つであり、1979 年日本の左右田健次教授らにより Trichoderma viride Y 244-2 株由来の L-リジン-α-オキシダーゼが初めて見つけられた[4, 5]。彼らは、L-リ ジンの α-アミノ基から起こる酸化的脱アミノ化反応の触媒機構について初めて議論 した。その後、スペインのグループにより Marinomonas mediterranea 由来の L-リジ ン-ε-オキシダーゼが同定されており[6]、現在まで L-リジンオキシダーゼの研究が活 発に行われている。L-リジンオキシダーゼの反応メカニズムを図 2-1 に示した。酸 化的脱アミノ化反応が起きる場所によって、α-オキシダーゼと ε-オキシダーゼに分 類されるが、一般的に α-アミノ酸側で反応が起こる場合が多いため、通常の L-リジ ンオキシダーゼは L-リジン-α-オキシダーゼを意味する。この反応で、L-リジン-α-オ キシダーゼは酸素分子が消費されてアンモニア(ammonia)、過酸化水素(hydrogen peroxide)、2-keto-6-aminocaproic acid が生成され、自発的に環形誘導体である Δ1 piperidine-2-carboxylate に変換される。L-リジン-ε-オキシダーゼは ε-アミノ基で反応 が起こるため 2-amino-6-semialdehyde adipic acid を生成する。これらの L-リジン オ キシダーゼを用いて L-リジンを検出·定量する方法やバイオセンサーへの応用などが 研究されている[7-10]。 89 ・Pseudomonase sp. AIU 813 由来の L-アミノ酸オキシダーゼ/オキシゲナーゼ(L-AAOd/Og) 最近の研究により、新たな L-アミノ酸オキシダーゼが発見された。共同研究者の 岩手大学の磯部教授らは、土壌細菌 Pseudomonas sp. AIU 813 株から新しい L-アミノ 酸オキシダーゼ(L-AAOd/Og)を同定してその機能解析を行った[11]。リジンを窒素源 とした培地で生産した L-AAOd/Og は、L-リジン、L-オルニチン(L-Ornithine)、L-ア ルギニン(L-Arginine)のみに対する活性を持っており、他の L-アミノ酸や D-アミノ 酸に対する活性は示さなかった。L-AAOd/Og の各基質とその基質特異性を図 2-2(a)、 表 2-1 に示した。L-AAOd/Og は、L-リジンに対する活性を 100%としたときに、Lオルニチン、L-アルギニンの活性は各々31%、6%であり、また L-リジンへは pH 5 9.5 の広い範囲での pH 安定性を持つものの L-オルニチンや L-アルギニンは pH 6 以 下の酸性側でその活性を失っていた。これらの結果から、この L-アミノ酸オキシダ ーゼは主に L-リジンを酸化する L-リジン-α-オキシダーゼであることが分かった。ま た表 2-1 の結果から L-AAOd/Og は、他の L-リジン-α-オキシダーゼより基質特異性 が高く、より広い pH 範囲で活性を持つことから、目的のアミノ酸のみを検出、定 量するために最適な新規酵素であると考えられる。一方、彼らが行った N 末端配列 解析の結果、L-AAOd/Og の N 末端配列は他の LAAO family とは異なっており、P. fluorescens Pf-5 由来 flavin-containing amine oxidase [12]、P. brassicacearum NFM421 由 来 oxidoreductase [13]、P. fluorescens Pf0-1 由来 tryptophan 2-monooxygenase [14]と類似 であることが分かった。Yamauchi らの報告(1975 年)によると、オキシダーゼ活性を 持ってない P. fluorescens 由来 L-リジンモノオキシダーゼは、p-chloromercuribenzoate (pCMB) のような スルフヒドリル試薬(Sulfhydryl reagent)によってそのスルフヒドリ ル基が修飾されて L-リジンモノオキシゲナーゼの活性が阻害されるとともに L-リジ ンオキシダーゼの活性が引き起こされた[15, 16]。ここに着眼した共同研究者の富山 県立大学の浅野教授らは、pCMB 修飾によって L-AAOd/Og のオキシダーゼ活性が約 5 倍以上上昇することを示した。また、L-AAOd/Og がモノオキシゲナーゼとしての 活性も持っており(図 2-2(b))、L-AAOd/Og のスルフヒドリル基の修飾はモノオキシ ゲナーゼ活性を阻害することも明らかにした。また、彼らは高い保存性を持つ 5 つ のシステイン残基に飽和変異(Saturation mutagenesis)を導入し、オキシダーゼ活性が 強化された変異体酵素 C254I を見出した[D. Matsui et. al., Unpublished data]。この変 異体酵素 C254I は、Wild type L-AAOd/Og よりオキシダーゼ活性が高く、pCMB によ る活性の変化が見られない酵素である(図 2-3)。これらの結果から 254 番のシステイ ン残基は、オキシダーゼとモノオキシゲナーゼ活性の変換に関与する非常に重要な 残基であると予測される。 このように L-AAOd/Og はオキシダーゼ活性とオキシゲナーゼ活性を同時に持つ 新規二機能性酵素であり、構造既知の LAAO family 及び Flavin monooxygenase 90 (FMO) family とのアラインメント·分子系統樹によると、両ファミリーとは違うとこ ろに位置していたため、違う祖先を持つ新規の酵素群である可能性が示唆された(図 2-4)。また、スルヒドリル試薬による活性の転換、そこに関与する残基の発見によ る変異体酵素の作製など、蛋白質工学分野でも非常に興味が深い酵素であると考え られる。本研究では、L-リジンに高い基質特異性を持つ新規 L-アミノ酸オキシダー ゼ/オキシゲナーゼ、L-AAOd/Og の構造解析を行い、本酵素が持つ新規構造及び基 質反応機構を明らかにすることを目的とする。また、変異体酵素 C254I の構造解析 を通し、モノオキシゲナーゼ活性からオキシダーゼ活性への活性変換の鍵であると 考えられる 254 番のシステイン残基の構造を明らかにし、活性変換機構の構造生物 学的解明を試みた。 91 実験の目的と方法 ・試薬 本研究で用いた試薬は特に記さない限り和光純薬工業株式会社、SIGMA-aldrich 社 のものを使用した。 ・実験器具 吸光光度計 V-550/560 (JASCO)、Benchmark Plus (BIO-RAD)、Nanodrop ND-1000 を 活性測定などに使用した。超音波破砕機は BIORUPTOR UCW-201 型 (COSMO BIO)、 SONIFIER 250D (BRANSON)を使用した。 ・培地 本研究ではすべて大腸菌を用いて実験を行ったため、特に表記しない限り LB 培 地(DIFCO)を用いた。すべての LB 培地はオートクレーブ滅菌を使った。試験官培養 は、水槽式インキュベーター(TAITEC)を使用し、大量培養の場合は、低温恒温槽付 回転式振とう培養機(高杉製作所)を使用した。抗生物質を添加する場合は、プラス ミドの種類に応じてアンピシリン 100 μg/ml、またはカナマイシン 50 μg/ml、クロラ ムフェニコール 100 μg/ml を添加して培養を行った。 ・遺伝子操作 Molecular Cloning 及び Current Protocol in Molecular Biology に記されている方法に 従った。 ・発現ベクター·菌株 本研究で取り扱ったリジン酸化酵素は、岩手大学農学部応用生物化学課程の磯部 公安教授らのグループより遺伝子がクローニングされ、大腸菌による発現ベクター は富山県立大学工学部生物工学研究センターの浅野泰久教授らのグループにより作 成された。提供していただいたリジン酸化酵素のプラスミドは、pET-15b のマルチ クローニングサイトに 5'側は Nde I で、3’側は Xho I で組み込まれ、N 末端に His-Tag が付加されている(図 2-5)。プラスミドの保持用の菌株は E. Coli XL10-GOLD 92 (Stratagene)を用い、発現用菌株としては E. Coli BL21 (DE3)-Codon plus (Stratagene)を 用いた。 2.2 Native L-AAOd/Og の発現と精製 ・目的 本研究の主な目的である X 線構造解析のためにタンパク質の結晶化を行う。良質 の結晶を作るためには、高純度で大量のタンパク質が必要である。そのために、ま ず発現条件の検討を行い、最も大量に発現する条件を見つけ出して大量発現·精製系 の構築を行った。 ・大量発現の条件探索 大量に発現する L-AAOd/Og の最適な発現条件を探すために発現条件の検討を行 った。誘導温度(25°C, 30°C)、IPTG の終濃度(0.5 mM, 1 mM)、培養スケール(100 ml, 1.5 L)などを検討した。詳しい発現の条件及び操作法は、以下に示す。 ・組み換え L-AAOd/Og の大量発現 ① 組み替え L-AAOd/Og を発現用の大腸菌へ形質転換した。発現用の大腸菌株は、 E. coli BL21 (DE3)-CodonPlus を用いた。 ② ①で作成した組み換え大腸菌をグリセロールストックより、LB 培地 10 ml に植 菌し、37°C で一晩振とうで前培養を行った。 ③ 前培養した 10 ml の培養液を LB 培地 3 L (5 L はね付き三角フラスコに 1.5 L 入れ たのも 2 本)に植菌し、37°C で本培養を開始した。 ④ 約 4 時間後に、終濃度 0.5 mM の IPTG を添加し、25°C で 12 時間振とう培養し た。 ⑤ 培養液を回収して 8,000 rpm、10 分間の遠心で集菌した。 ・L-AAOd/Og の精製 93 大量培養した L-AAOd/Og を集菌し、その菌体を 150 mM の 50 mM HEPES–NaOH (pH 7.0)緩衝液で懸濁し、出力レベル 5.5、Duty 50%、Palse 1 sec で、25 分間超音波 破砕を行った。その後、12,000 rpm、45 分間の遠心で集菌した。上清の可溶性画分 のみを採取し、Minisart syringe filter 0.45 μm (Sartorius Stedim)を用いて微粒子を除去 した。 Ni-NTA Superflow column chromatography この精製の制御には、ペリスタポンプ(Econo gradient pump, Bio-RAD)を用いた。 ① C 16/20 column (GE Healthcare Bioscience)に Ni-NTA superflow resin を 5 ml 充填し、 50 mM HEPES–NaOH (pH 7.0)を用いて平衡化を行った。 ② サンプルを①に添加し、50 mM HEPES–NaOH (pH 7.0) + 100 mM Imidazole で非 吸着画分を溶出した。 ③ 50 mM HEPES–NaOH (pH 7.0) + 500 mM を用いて吸着画分を溶出した。 ④ 分離した吸着画分を Amicon Ultra-15 10K (Millipore) を用いて濃縮·脱塩を行った。 濃縮脱塩後のバッファーは 50 mM HEPES-NaOH (pH 7.0)を用いた。 MonoQ column chromatography 以降の精製の制御には、AKTA FPLC (Fast Protein Liquid Chromatography) System (GE Healthcare Bioscience)を用いた。カラムは MonoQ HR 10/100 GL (GE Healthcare Bioscience)を用いて、カラムの許容量を超えないようにサンプルを 1/3 ずつ、3 回に 分けて陰イオン交換カラムクロマトグラフィー精製を行った。 ·A buffer: 50 mM HEPES-NaOH (pH 7.0) ·B buffer: 50 mM HEPES-NaOH (pH 7.0) + 1 M NaCl ① 上記のサンプルを Ultrafree-MC 0.45 μm (Millipore)を用いて微粒子を除去した。 ② A バッファーでカラムを平衡化した。 ③ 2 ml 容量のサンプルループを用いてサンプルをカラムに吸着させ、A バッファ ーで非吸着画分を溶出した。 ④ A バッファーと B バッファーで 0 M から 1 M まで NaCl のグラジエントをかけ て吸着画分を溶出した。 94 ⑤ 280 nm の UV モニターリングやタンパク質の定量から、目的タンパク質が多く 含まれている画分を回収。分離した画分は Amicon Ultra-15 10K を用いて濃縮·脱 塩を行った。交換後のバッファーは 50 mM HEPES-NaOH pH 7.0 + 150 mM NaCl にし、その容量は 1 ml 以下になるようにした。 Gel filtration column chromatography ·Running buffer: 50 mM HEPES-NaOH pH 7.0 + 150 mM NaCl ① MonoQ カラムクロマトグラフィーで分離した吸着画分は Ultrafree-MC 0.45 μm を 用いて微粒子を除去した。 ② Hiload 16/60 Superdex 200 prep grade (GE Healthcare Bioscience)カラムを Running buffer で平衡化を行った。 ③ 1 ml 容量のサンプルループを用いてサンプルをカラムに吸着させ、Running buffer で溶出した。 ④ 280 nm の UV もモニターリングでタンパク質の分離を確認し、分離した画分を Amicon Ultra-4 10K で濃縮·脱塩を行った。 ・タンパク質定量 タンパク質の定量は、BCA (Bicinchoninic acid)法により行った。BCA working reagent (PIERCE)の A と B バッファーを 50:1 で混ぜた溶液とサンプルを 20:1 で混合 し、60°C で 30 分間反応した後、562 nm の吸光を測定した。標準タンパク質として は、BSA (Bovine Serum Albumin)を用いて 0、0.05、0.1、0.15、0.2、0.25、0.5 mg/ml の濃度で検量線を描いた。 2.3 L-AAOd/Og セレノメチオニン置換体の作製 ・目的 L-AAOd/Og とアミノ酸配列相同性がある構造既知の類似酵素の配列相同性が 22%以下で低いため、分子置換法による位相決定は困難であると判断した。そこで、 X 線の初期位相を決定するために L-AAOd/Og のセレノメチオニン置換体を作製し、 Single-wavelength anomalous dispersion (SAD) 法 や Multiple-wavelength anomalous 95 dispersion (MAD)法による位相の決定を目指した。セレンは λ=0.98Å 付近で異常分散 を起こすため、新規構造の位相決定に最もよく使われる方法である。 ・発現の宿主菌株及び大量発現 タンパク質のセレノメチオニン置換体の作製は、メチオニン要求性の大腸菌株を、 セレノメチオニンを含む最少培地で培養して行う。この方法では、メチオニン要求 性の菌株にタンパク質発現用のプラスミドを形質転換する必要があるため、LAAOd/Og の発現用プラスミド pET-15b を、メチオニン要求株である E. coli B834 (DE3)及び E. coli BL21 CodonPlus (DE3)-RIL-X 株に形質転換した。小スケールの発現 チェックにより後者の方で多く発現することを確かめ、E. coli BL21 CodonPlus (DE3)-RIL-X 株に形質転換したサンプルを使用した。 ① グリセロールストックより、LB 培地 10 ml に植菌し、37°C で一晩振とう培養し た。(前培養) ② 8,000 rpm、10 分間の遠心で集菌した。 ③ 菌体をセレノメチオニン培地で洗浄して LB 培地を除去した。(2 回繰り返し) ④ 1 L のセレノメチオニン培地**(アンピシリン 100 μg/ml)2 本に植菌し、37°C で 6 時間本培養を行った。 ⑤ 終濃度 0.25 mM の IPTG を添加し、20°C で一晩振とう培養でタンパク質を誘導 した。 ⑥ 培養液を回収して 8,000 rpm、10 分間の遠心で集菌した。 **セレノメチオニン培地組成 (1 L) コア培地(Wako) 23.5 g(オートクレーブ)、グルコース 10 g(別途オートクレーブ)、 MgSO4·7H2O 250 mg、FeSO4·7H2O 4.2 mg、Selenomethionine 50 mg (0.22 μm フィルタ ー滅菌)、Sigma KAO & Michaylak vitamin solution 10 ml ・精製 セレノメチオニン置換体の精製は、2.2 節で行った Native L-AAOd/Og の精製法と 同様に Ni-NTA Superflow column chromatography、MonoQ column chromatography、Gel filtration column chromatography の 3 段階の精製を行った。 96 2.4 Native L-AAOd/Og と L-AAOd/Og Se-Met 置換体の結晶化条件探索 ・目的 L-AAOd/Og の X 線構造解析を行うため、高分解能の回折を与える結晶を得ること が必要である。そのため、2.2、2.3 節で精製した Native 及び Se-Met 置換体のサンプ ルを用いて結晶化スクリーニングを行い、結晶化条件を探索した。 ・結晶化条件のスクリーニング L-AAOd/Og の結晶化に適するタンパク質の濃度を見積もるために、PCT (PreCrystallization Test, Hampton Research)キットを用いた結果、約 20 mg/ml 程度が適切な 濃度であることが分かった。 結晶化条件を探索するために、Sparse Matrix Screen 法による結晶化のスクリーニ ン グ を 行 っ た 。 結 晶 化 条 件 の ス ク リ ー ニ ン グ は 、 イ ン テ リ プ レ ー ト (Hampton Research)を用いて、シッティングドロップ蒸気拡散法により行った。使用したキッ トと結晶化の手順は以下に示す。 ·JCSG + Suite, JCSG Core 1 ~ 4 Suite (QIAGEN)、 ·Crystal Screen 1 & 2 (Hampton Research) ·Wizard 1 & 2, Precipitant Synergy (Emerald BioSystems) ·Crystal Screen Cryo & lite (Hampton Research): Sparse matrix の derivative ·SaltRX, PEG/Ion 1 & 2 (Hampton Research): 塩と PEG の組み合わせ ① インテリプレートの各ウェルにスクリーニングキットの各溶液を 70 μl ずつ分注 し、リザーバー溶液とした。 ② インテリプレートの各ドロップにサンプルとリザーバー溶液を各々0.5 μl ずつ混 ぜた。 ③ Crystal Clear Sealing Tape (Hampton Research)を用いてインテリプレートを密閉し、 20°C に静置した。 ・結晶化条件の精密化 97 結晶が得られた条件について、さらに大きく質が良い結晶を得るため、沈殿剤濃 度、pH、タンパク質濃度、結晶化方法などについて精密化を行った。 2.5 X 線回折データ測定 ・目的 2.4 節で得られた結晶について、X 線を照射して回折イメージを測定した。放射線 による結晶の損傷を防ぐために、95 ~ 100 K の窒素ガスでフラッシュクーリングし た。また、低温での結晶の凍結を防ぐため、抗凍結剤である 20%グリセロールを含 む結晶化溶液に結晶を浸透させた後、Native 結晶は 1.0 Å の波長で、Se-Met 置換体 結晶は 0.97921 Å でデータ測定を行った。 ① 3 Well Spot Plate (Hampton Research)のウェルに結晶化リザーバー溶液を 100 μl 入 れた。各ウェルの抗凍結剤の濃度を少しずつ上げ、終濃度を 20%にした。 ② 結晶化プレートから結晶を慎重にすくい、ウェルのリザーバー溶液に回収した。 準備した 0、10、20%のグリセロールを含む結晶化溶液にステップワイズ法で順 に移して結晶に抗凍結剤の濃度を上げた。 ③ 結晶をクライオループ(Hampton Research)にマウントし、100 K のクライオスト リームの中に設置して瞬間的に冷却し、X 線を照射して回折データイメージを 測定した(図 2-6)。 **X 線回折データ測定は、つくば市·高エネルギー加速研究機構(KEK)の放射光施設 Photon Factory (PF)実験ステーション BL1A、BL5A、NW12A で行った。 2.6 初期構造決定及び構造精密化 ・L-AAOd/Og セレノメチオニン置換体の MAD 測定 セレノメチオニン置換体 L-AAOd/Og の回折データ測定は、KEK-PFAR NW12A で 行った。データ測定は 2.5 節の操作を従い、0.9790 ~ 0.9800 Å の連続した波長の X 98 線を照射した。XAFS 測定により、異常分散効果の最も大きい波長から MAD デー タの測定波長(Peak: 0.97921 Å、Edge: 0.97932 Å、Remote: 0.96409 Å)を決定した後、 その異なる 3 波長の X 線を照射して MAD データを測定した(図 2-7)。 ・位相位相決定と構造精密化 処理した MAD データを用いて PHENIX[17]の AUTOSOL で初期位相を決定しようと したが、実際の結果では MAD のデータより SAD を用いた時の結果が良かったため、 最終的には SAD データを用いることにした。得られた電子密度を用いて CCP4[18] の Buccaneer でペプチド鎖の自動構築を行い、もう 1 回 Arp/wArp[19]を用いて自動 モデル構築を行った。Molrep[20]により Native データとの分子置換を行い、その後 Refmac5 で自動精密化を行った。Coot を用いてペプチド鎖の手動構築及び水分子処 理など細かい精密化を行い、再度 Refmac5 により自動精密化を行って最終構造を得 た。 ・構造解析 本研究で得られた L-AAOd/Og の全体構造及びドメインなど構造の新規性や有意 性を解析するために Dali Server (http://ekhidna.biocenter.helsinki.fi/dali_server/)を用いた。 基質及び酸素の経路(チャンネル)を解析するために CAVER 2.1 を用いた[21]。LAAOd/Og の立体構造に関するすべての図は PyMOL を用いて描いた[22]。その他に 解析に用いたサーバやプログラムなどを以下に示す。 ·ExPASy Proteomics Server (http://www.expasy.ch/cgi-bin/protparam) ·Matthews Probability Calculator (http://ruppweb.dyndns.org/mattprob/) ·MOLPROBITY (http://molprobity.biochem.duke.edu/) ·Cremer-Pople parameter calculator (http://asfushi.hp.infoseek.co.jp/) 2.7 基質複合体の構造解析 ・目的 基質認識部位の同定及び基質との結合様式を明らかにするため、本酵素の基質で ある L-Lysine との複合体の構造解析を行った。L-AAOd/Og に対して各々31%、6% の活性を示す L-Ornithine、L-Arginine との複合体の構造解析も同時に試みた。 99 ・ソーキング法 2.4 節で得られた Native L-AAOd/Og の結晶についてソーキング法による複合体の 構造解析を試みた。用意した抗凍結剤を含むリザーバー溶液に終濃度 10 mM になる ように基質を添加し、2.5 節と同様に結晶に浸透させた後 X 線回折データを測定し た。 ・共結晶化 L-Lysine、L-Ornithine、L-Arginine の 3 種類の基質をそれぞれ 5 - 10mM となるよ うに加えて共結晶化を行い、基質が結合した状態の結晶を得ることを試みた。実験 の方法は、2.4 節の結晶化条件の探索と同様に行った。 2.8 変異体酵素 C254I の構造解析 ・目的 上述のように、L-AAOd/Og の変異体酵素 C254I は L-AAOd/Og のオキシゲナーゼ 活性を完全にオキシダーゼに転移し、pCMB による修飾を受けないように作られた 変異体である。変異体酵素 C254I の結晶構造解析により、活性転移のメカニズムを 構造的観点で明らかにするために、本実験を行った。L-AAOd/Og C254I の遺伝子サ ンプルは、富山県立大学の松井大介博士から提供して頂いた。 ・L-AAOd/Og 変異体 C254I の大量発現 小スケールの発現条件検討により L-AAOd/Og C254I の発現条件を決定した。発現 条件の検討では、培養温度(37°C、25°C)、誘導温度(37°C、25°C、20°C)、IPTG 濃度 (0.1 ml、0.5 ml、1 ml)、発現菌株(E. coli BL21 (DE3)-CodonPlus、E. coli BL21 (DE3)、 E. coli C43 (DE3))などを検討した。 ① 変異体 C254I のプラスミドを発現用の大腸菌へ形質転換した。発現用の大腸菌株 は、E. coli BL21 (DE3)-CodonPlus を用いた。 100 ② ①で作製した組換え大腸菌をグリセロールストックより、LB 培地 10 ml に植菌 し、37°C で 1 晩振とうで前培養を行った。 ③ 前培養した 10 ml の培養液を LB 培地 3 L (5 L はね付き三角フラスコに 1.5 L 入れ たのも 2 本)に植菌し、20°C で本培養を開始した。 ④ 約 12 時間後に、終濃度 1 mM の IPTG を添加し、20°C で 12 時間振とう培養した。 ⑤ 培養液を回収して 8,000 rpm、10 分間の遠心で集菌した。 ・L-AAOd/Og 変異体 C254I の精製 L-AAOd/Og C254I の精製は Ni-NTA Superflow column chromatography、Gel filtration column chromatography の 2 段階の精製を行った。詳しい精製方法については、2.2 節 と同様に行った。 ・L-AAOd/Og 変異体 C254I の結晶化条件の探索 まず 2.12 節の Native L-AAOd/Og の結晶化条件探索の結果に従って、Native の結 晶が作られた条件で結晶化を行った。その条件を中心とし、タンパク質濃度、塩濃 度、PEG 濃度、pH などの条件精密化を行った。また、以下に示すキットを用いて 更なる結晶化条件の探索を行った。 ·PEG/Ion 1 & 2 (Hampton Research) ·JCSG + suite (QIAZEN) 2.9 pCMB 修飾 L-AAOd/Og の構造解析 ・目的 L-AAOd/Og は pCMB 修飾によって本来持つオキシダーゼ活性を失い、微弱なオキ シダーゼ活性が増大する特徴がある[15]。本実験では、2.8 節の変異体酵素 C254I の 構造解析と共に Native L-AAOd/Og が pCMB の修飾を受けることによって起こる構 造上の変化を明らかにするため、pCMB 修飾 L-AAOd/Og の構造解析を行った。 ・ソーキング法 101 2.4 節で得られた Native L-AAOd/Og の結晶についてソーキング法による pCMB 修 飾 L-AAOd/Og の構造解析を試みた。0.2 mM の pCMB 処理でほぼ最大のオキシダー ゼ活性を示すため(松井大介博士のデータより)、抗凍結剤を含むリザーバー溶液に 終濃度 0.2 mM になるよう pCMB を添加し、2.5 節と同様に Native L-AAOd/Og の結 晶に浸透させた後 X 線回折データを測定した。 ・共結晶化 サンプルに pCMB を 0.2 - 1mM となるように加えて共結晶化を行い、pCMB 修飾 の L-AAOd/Og の結晶を得ることを試みた。実験方法は 2.4 節の結晶化条件の探索と 同様に行った。 2.10 L-AAOd/Og の諸性質の解析 ・目的 L-AAOd/Og の基本的な性質は、野生型 L-AAOd/Og(Pseudomonas sp. AIU 813 株か ら精製したもの)について解析されている[11]。本節では大腸菌を用いて発現した組 み換え酵素 L-AAOd/Og の基本的な諸性質について調べた。 ・分子量測定(4 次構造予測) 分子量測定及び 4 次構造予測は、 Hiload 16/60 Superdex 200 pg カラムを用いたゲ ルろ過カラムクロマトグラフィーにより行った。分子量マーカーとして、 Tyroglobulin (669 kDa)、β-Amylase (200 kDa)、Albumin (66 kDa)、Ovalbumin (45 kDa)、 Trypsin inhibitor (21 kDa)を用いた。 ① Hiload 16/60 Superdex 200 pg カラムを 50 mM HEPES-NaOH pH 7.0 + 150 mMNaCl で平衡化を行った。 ② 精製·濃縮したサンプルを 50 mM HEPES-NaOH pH 7.0 + 150 mM NaCl 適切な濃度 で希釈してカラムに吸着させ、その保持時間を測定した。 ③ 上記の分子量マーカーに関しても同様の操作で保持時間を測定した。 102 ・L-AAOd/Og の活性測定法 L-AAOd/Og の基本的な活性測定は、基質の酸化時に発生する過酸化水素をペルオ キシダーゼと反応させるカップリング反応として行った(図 2-8)。酵素反応液の組成 は、0.1 M Potassium phosphate pH 7.0、0.6 mM 4A·A (4-Aminoantipyrine)、1.94 mM TOOS (N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3methylaniline sodium salt dehydrate)、0.2 mM FAD (Flavin-adenine dinucleotide)、12.7 units peroxidase、60 mM Substrate である。 ① 酵素反応液 1 ml をキュベット(SARSTEDT)に分注し、30°C 保たれている分光光 度計内に設置して 10 分間プレインキュベートを行った。 ② その後、キュベットは設置したまま酵素液 10 μl (1 ~ 5 mg/ml)添加、ピペットで 混合した。 ③ 波長 555 nm における吸光度の増加を測定した。 ・吸収スペクトル 組み換え L-AAOd/Og は高濃度になると黄色を呈した。この黄色は L-AAOd/Og の 補因子として結合している FAD が原因である。一般的に補因子である FAD は酸化 状態で酵素に結合しており、還元状態になると FADH2 になる。本節では、FAD がも つ 2 つの固有波長(350 ~ 500 nm)での吸光度を計って酵素の酸化還元状態を判別する 実験を行った。L-AAOd/Og に基質(L-Lys)、pCMB、還元剤などを添加し、好気·嫌気 の条件で各々の吸収スペクトルを測定した。 ① L-AAOd/Og を 50 mM HEPES pH 7.0 を用いて 0.4 mg/ml となるように希釈し、総 量 400 ml をキュベットに分注した。 ② pCMB 存在·非存在下で 1 mM L-Lys を添加し、よく混合した。 ③ 波長 200 nm ~ 700 nm の吸収スペクトルを測定した。 ④ 嫌気条件では、1 mM L-Lys を添加した後に 0 min、30 min で各々測定した。反 応時の嫌気条件は、反応液に N2 ガスをパージすることによって整えた。 103 結果と考察 2.11 組換え L-AAOd/Og の精製と諸性質の解析 ・大量発現と精製 小スケールで行った発現条件の探索の結果から誘導温度 25°C での発現量が最も多 かったため、25°C での誘導でタンパク質の大量発現を行った。L-AAOd/Og の大量 発現·精製の結果を図 2-9、表 2-2 に示す。アミノ酸配列による L-AAOd/Og の推定分 子量は 62 k であったが、SDS-PAGE の結果では 55 k の付近でバンドが見られた。由 来菌である Pseudomonas sp. AIU 813 株から精製した酵素も同様に 55 k を示したため [11]、組換えによるエラーではないと考えられる。本酵素のアミノ酸配列からの分 子量計算と SDS-PAGE で見られる分子量が異なる理由は不明である。3 L の培養か ら約 30 mg のタンパク質を得ることができた。SDS-PAGE の結果では、3 つの精製 ステップを経ることでほぼ単一バンドとなっていることが分かったが、目的バンド 以外の一部の余計なバンドに関しては、ゲルろ過の UV モニターリングの結果から その余計なピークに相当するピークが観察されなかったため、そのまま結晶化スク リーニングを行った。 ・分子量測定/4 次構造予測 L-AAOd/Og の野生型酵素(Native L-AAOd/Og)、変異体酵素(C254I)の溶液中の分子 量を測定し、その 4 次構造を予測した。標準タンパク質分子量マーカーの溶出パタ ーン(図 2-10(a))、マーカーと L-AAOd/Og の溶出パターンの関係を図 2-10(b)にプロ ットした。溶出容量から見積もられた L-AAOd/Og の分子量は約 219 k であった。ア ミノ酸配列による L-AAOd/Og の分子質量は約 62 k であったため、L-AAOd/Og は溶 液中で 4 量体を形成していることが示唆された。また、変異体酵素の溶出パターン では、もう 1 つのピーク(peak 2)が検出され、その分子量を調べたところ、約 1430 k であり、約 24 量体の L-AAOd/Og に相当していた。この結果から、L-AAOd/Og の変 異体酵素は溶液中で部分的に不安定な不規則重合を形成していると考えられる。 ・吸収スペクトル (酵素の分光学的性質) 好気条件で行った L-AAOd/Og の吸収スペクトルの結果を図 2-11 に示した。LAAOd/Og はリジンとの反応により還元状態になるため FAD の固有波長でのスペク 104 トルが減少すると予測される。実際 1 mM のリジンを添加したとき、野生型の酵素 よりスペクトルが若干減少するものの大きな差は見られなかった(図 2-11(b))。pCMB の処理は、L-AAOd/Og のオキシダーゼ活性を 5 倍以上上昇するという報告に基づき、 pCMB 存在下でリジンとの反応を測定したが、結果に変化は無かった(図 2-11(c))。 好気条件では、空気中の酸素の影響があるため酸化還元の判断が難しいと考えて嫌 気の条件で新たに実験を行った(図 2-12)。基質の濃度、反応時間を変化して FAD の スペクトルを測定した結果、高濃度のリジンを添加したときに FAD の固有スペクト ルが減少していることが確認された(図 2-12(b))。これらの結果から、L-AAOd/Og は リジンとの反応によってフラビンが還元され、酵素が還元状態になることが確かめ られた。 2.12 L-AAOd/Og の結晶化条件の探索結果 精製した L-AAOd/Og を用いて結晶化条件のスクリーニングを行った。PCT(PreCrystallization Test)の結果から結晶化に適したタンパク質の濃度は約 20 mg/ml に設定 し、2.4 節に記述したように Crystal Screen 1 & 2、Structure Screen 1 & 2 (HR)、JCSG + suite (QIAZEN)、Precipitant Synergy (EB)などのキットを用いてスクリーニングを行 った。多数の条件で結晶が作られたが、外見的に質が良い結晶は少なかった。その うち一番綺麗な結晶(図 2-13(a))が作られた条件を以下の方に示す。 ·0.1 M Sodium acetate pH 4.6, 8 % Polyethylene glycol 4000 Se-Met 置換体 L-AAOd/Og の結晶化は、まず Native L-AAOd/Og の結晶が作られた 条件を中心に行った。しかし、ほとんどの条件で結晶は成長せず、再現性を得るこ とはできなかった。特に、一番綺麗な Native L-AAOd/Og の結晶が得られた 0.1 M Sodium acetate pH 4.6, 8 % Polyethylene glycol 4000 の条件でも再現性が見られなかっ たため、キットの結晶化溶液の pH を確認した結果、4.6 であるはずの溶液の pH が 中性に近く変わっていた。pH を中性に近くして、結晶化条件の最適化を行ったが、 結晶は作れるものの質が良い結晶を得ることはできなかった。そのため、もう 1 回 Se-Met 置換体 L-AAOd/Og について結晶化条件のスクリーニングを行った結果、 PEG3350 が 含 ま れ た 条 件 で 結 晶 が よ く 作 ら れ る 傾 向 が あ る こ と が 分 か っ た 。 PEG/Ion 1 & 2 (HR)キットは PEG の種類を PEG3350 に固定し、塩濃度や PEG3350 の 濃度を組み合わせたキットであるため、PEG/Ion 1 & 2 キットを使用することによっ て、沢山の条件で質が良い結晶を得ることができた。その中で一番質が良い結晶(図 2-13(b))が得られた条件を以下の方に示す。 ·0.02 M Citric acid, 0.08 M, Bis-Tris propane pH 8.8, 16% PEG 3350 105 ソーキング法による基質複合体構造を得るためには、Native の結晶を沢山作る必 要がある。そのために一番最初に Native 結晶を得た 0.1 M Sodium acetate pH 4.6, 8 % Polyethylene glycol 4000 の条件や pH を中性に変えた条件で結晶化を試みたが、質が 良い結晶を得ることはできなかった。その後、PEG3350 が含まれる条件での最適化 により、良質の綺麗な結晶(図 2-13(c))を得ることができた。その条件を以下の方に 示す。 ·2% Tacsimate, 14.3% PEG3350, 0.1 M Tris-HCl pH 8.0 ·0.15 M DL-Malic acid pH 7.0, 20 % PEG3350 上記の結晶はすべてシッティングドロップ蒸気拡散法により、20°C で 1~2 週間放 置することによって作られた。これらの結晶を用いて X 線回折データ測定を行った。 2.13 構造解析の結果 高エネルギー加速器研究機構(KEK)の放射線施設 Photon Factory (PF)実験ステーシ ョン BL1A、BL5A、NW12A を利用し、得られた結晶に対して X 線回折データの測 定を行った。回折データについての解析及び処理は、HKL2000 を用いて行った。 Native L-AAOd/Og の最大分解能は 1.9 Å であり、空間群は P21212、Native 結晶の単 位胞のパラメーターは a=118.926、b=141.353、c=75.970 (Å)、α=β=γ=90°であった。 VM=2.62 Å3/Da、Vsolvent=53.12%であり、非対称単位中に 2 分子の L-AAOd/Og が入っ ていた。X 線回折データの測定結果を表 2-3 に示した。 ・初期構造の構築 Se-Met 置換体の結晶は、Native の結晶とは違う結晶系を示した。最大分解能は 2.2 Å であり、格子定数は a=74.180、b=101.136、c=151.730 (Å)、α=β=γ=90°、空間群 は I222 であった。この結晶の非対称単位中には 1 分子の L-AAOd/Og が入っていた。 MAD 測定に用いた波長は、XAFS の測定結果より、Peak 0.97921 Å、Edge 0.97932 Å、 Remote 0.96409 Å とした。3 つの波長で測定した MAD データを用いて X 線の初期 位相を決定しようとしたが、SAD データ(Peak の 1 波長のみ)を用いたときの結果が より良かったため、SAD データを用いてプログラム PHENIX の AutoSol より初期位 相を決定した。L-AAOd/Og に含まれるメチオニン残基(セレノメチオニンに置換)18 個を上回る、25 個のサイトを用いて位相の計算を行った。位相の確からしさを示す Figure of merit (FOM)は、計算された位相と真の位相のずれを示すパラメータで、1 に近いほど真の位相と差が小さいことを示す。AutoSol により計算された初期位相で 106 は、FOM が 0.437 となった。AutoSol では L-AAOd/Og の 560 残基のうち 504 残基の ペプチド鎖を構築したが、ところところ 2 次構造が組まれておらず不完全な構造を とっていた。そこで、CCP4 の BUCCANEER を用いて自動モデル構築を行った結果、 548 残基のペプチド鎖が自動構築されることによって、部分的に不完全であった 2 次構造がうまく構築された。BUCCANEER は低分解能およびノイズの多い電子密度 データを元にしてモデル構築を行うプログラムである。その後 ARP/wARP を用いて もう 1 回自動モデル構築を行うことにより、99%以上のペプチド鎖が構築された。 ・Native L-AAOd/Og の構造解析 Native L-AAOd/Og の精密化及び構造解析は、Se-Met 置換体結晶の回折データで構 築した初期構造を基にして、Native 結晶の回折データを分子置換することによって 行った。分子置換は Molrep を用いて行い、その後 Coot による手動モデル構築や Refmac5 による精密化によって、最終的に R-factor/Rfree=20.9/25.3%の構造を得ること ができた。精密化の結果を表 2-3 に示した。 ・基質複合体の構造解析 L-Lys、L-Orn、L-Arg との複合体構造解析のため KEK PF BL-5A にて、波長 1.0000 Å で X 線回折データの測定を行った。最大分解能は各々2.00、2.2、2.6 Å で あり、P21 の同じ空間群を示した。これら結晶の非対称単位中には 4 分子の LAAOd/Og が入っており、4 次構造予測の結果(2.11 節)の通り水溶液中で四量体とし て存在することと一番近い構造をとっていることが分かった。基質複合体の構造解 析は、Native L-AAOd/Og の構造を鋳型にして Molrep を用いた分子置換より行われ た。基質結合部位で見られた各基質の電子密度マップに従い、Coot での手動構築に て基質の構造を明らかにした。詳しい基質の結合様式に関しては 2.14 節で論じる。 Refmac5 により自動精密化された各構造の R-factor は 27.6, 21.0, 22.2%であるが、基 質複合体の構造精密化はまだ未完成であるため、さらに精密化を続ける必要がある。 現在まで精密化した結果については表 2-3 に示した。 2.14 構造に関する考察 ・全体構造 107 L-AAOd/Og の全体構造は、Native L-AAOd/Og の結晶では二量体を、基質複合体 の結晶では四量体構造を取っていた(図 2-14)。2-11 節でのゲルろ過クロマトグラフ ィーを用いた分子量測定の結果では四量体であると予測されたため、四量体の全体 構造を基本として二量体構造と比較しながら議論する。図 2-15 には L-AAOd/Og の 分子表面図を示した。二量体 Native L-AAOd/Og の分子表面図を横からみると、2 つ の分子は「⊏」のような形をしていた。つまり片方の正面から見たら長さ約 55 Å のト ンネルが形成されているが、180°回転した構造ではそのトンネルが見えなかった(図 2-15(a))。四量体の分子表面図では、4 つの分子の間を通す十字のトンネルが形成さ れていた(図 2-15(b))。これらのトンネルによる基質及び酸素の出入りに関しては本 節の「基質及び O2 チャンネルで詳しく述べる。 L-AAOd/Og の各単量体は 19 本の α-helix と 19 本鎖の β-strand を含んでおり、3 つ のドメインから構成されている。補因子としての FAD が結合している FAD binding domain (S38-L83, G264-T358, G475-K538)、基質が結合する Aromatic cage (W516, Y473)が形成されている Substrate binding domain (K12-G37, R84-E147, Q258-G263, R359-K474, T539-D560)と両ドメインの間に存在する Helical domain (K148-H257)から なる構造をしていた(図 2-16)。FAD binding domain は 8 本の α-helix と 7 本鎖の βstrand から形成されており、FAD の結合部位を含んでいた。アミノ酸配列上、お互 い離れたところに位置している β1、β12、β19 が同じ β-sheet 上で連続的に並んでい ることなど複雑な構造をとっているのが特徴的である。基質触媒部位をもつ Substrate binding domain は 2 つの α-helix を貫通する 4 本の β-strand を中心として、 12 本鎖の β-strand と 4 本の α-helix により構成されていた。また、β16 と α16 を繋ぐ ループの配列(M419-H428)のマップが欠損していた。Helical domain は 7 本の α-helix のみで構成されたドメインで、他の 2 つのドメインの間で構造の安定化に寄与する 部分と考えられる。 ・類似構造の検索 構造既知タンパク質(Protein Data Bank に登録されたタンパク質の構造)と LAAOd/Og の構造的類似性を調べるため、Dali server を用いた[23]。Calloselasma Rhodostoma 由 来 L-amino acid oxidase (1F8R 、 Z-score: 35.2 、 RMSD: 2.3)[24] 、 Psuedomonas sp. P-501 由来 L-phenylalanine oxidase (2YR4、Z-score: 33.5、 RMSD: 3.0)[25] 、 Homo sapiens 由来 Monoamine oxidase (2BXR 、 Z-score: 30.7 、 RMSD: 3.7)[26]、Homo sapiens 由来 Lysine-specific histone demethylase 1 (3ABT、Z-score: 27.5、 RMSD: 3.7)[27]などの類似構造を持つ酵素がヒットした(図 2-17)。Dali server の結果 で上位にランクされたの酵素はほとんど LAAO (L-amino acid) family および FMO (Flavin monooxygenase) family に属しているため、L-AAOd/Og は両酵素の構造的特徴 を持っていると考えられる。また両ファミリーは FAD を補酵素として使用する酵素 108 群であるため、L-AAOd/Og の FAD binding domain はのこれらの酵素と非常に高い類 似性を示していた。Substrate binding domain、Helical domain のみの類似構造を調べ るため PDBeFold server[28]、Dali server を用いた結果、全体構造の類似性とほとんど 変わらなく、LAAO family に属する酵素が上位にランクされた。 ・活性中心及び基質複合体構造 L-AAOd/Og の 活 性 中 心 に は 、 FAD の Flavin ring 、 Trp516 と Phe473 に よ り Aromatic cage が形成されている(図 2-18)。この部分は Monoamine oxidase が持つ Aromatic cage と非常に似ており、Monoamine oxidase では 7.8 Å の幅を持つものに対 し、本酵素では 7.9 Å の幅を持つ[29]。FAD の Flavin ring は Aromatic cage 側に少し 曲がっているが、基質が flavin ring の曲がった部分である N5 原子に酸素原子を提供 して反応が起き易くなっていると予測される。この基質結合部位は、Aromatic cage 以外にも Trp235、Trp418、Phe416 などの疎水性残基が位置しており、疎水性ポケッ トを形成していた。L-AAOd/Og の活性転換に重要な役割をしていると考えられる 254 番目のシステイン残基は Aromatic cage を形成する Trp516 のすぐ隣に存在してい る(図 2-18)。 3 つの基質複合体の構造では、L-AAOd/Og の基質である L-Lys、L-Orn、L-Arg が 各々Aromatic cage 付近の基質認識部位に結合していた。各基質の|Fobs| - |Fcalc|電子密 度マップと基質認識残基を図 2-19 に示した。3 つの基質の結合様式は大きな違いは なかったが、 L-Orn、L-Arg に対しての活性が L-Lys に比べてかなり低いため(30%, 6%)[11]、比較的長時間ソーキングを行ったことにもかかわらず、その結合が見えな い Chain があった。基質のアミノ基(-NH2)は Asp238 や 2 つの水分子と、カルボキシ ル基(-CO2-)は Arg102 と相互作用をしていた。 一方、電子伝達タンパク質間の酸化還元状態依存的な親和性調節機構は、昔から 多くの研究者の興味の的であり、特に電子伝達機構での補酵素として作用する FAD については詳しく研究されている[30]。千田らは、Acidovorax sp. strain KKS102 由来 芳香環水酸化ジオキシゲナーゼのコンポーネントのうち NADH 依存性フェレドキシ ン還元酵素(BphA4)の 4 つの酸化還元状態を明らかにし、その構造変化を同定した (図 2-20(a))。BphA の FAD は進行に従い、酸化型のときには平面であったイソアロ キサジン環が、二原子還元型のときには N5、N10 を中心に曲がっており、一電子還 元型ではイソアロキサジン環が捻れた構造をとっていた。この捻れ構造は、再酸化 により解消し、イソアロキサジン環は平面構造に戻る(図 2-20(a)③)。それに基づき、 本研究で得られた L-AAOd/Og の Native 型の構造と Lys 複合体型の構造を比べてみ たが、結晶化のときに基質を添加することによって結晶の色は黄色から白に薄くな るものの、構造上の FAD のイソアロキサジン環やリビチル鎖の有意差は見られなか 109 った(図 2-20(b))。Rhodococcus opacus LAAO は基質と反応することによって還元され るが[31]、本実験では結晶中での酸化型中間体をしっかり取れなかったか、いくつ かの酸化還元状態が混在しているか、あるいは BphA4 より低分解のデータであるた め、その構造変化が見られなかった可能性があると考えられる。 ・他の構造既知の類似構造との比較 Dali server を用いた類似構造探索で見つけられた L-AAOd/Og と類似構造を持つ Calloselasma Rhodostoma 由来 L-amino acid oxidase (crLAAO)、Psuedomonas sp. P-501 由来 L-phenylalanine oxidase (PAO)との活性中心を比べてみた(図 2-21)。両酵素とも 活 性 部 位 を 認 識 す る 重 要 な 残 基 は ほ と ん ど 保 存 さ れ て い た 。 Calloselasma Rhodostoma 由来 crLAAO は、Substrate binding domain と Helical domain の間に 25 Å の長いトンネルが形成しており、そこに 3 つの o-aminobenzoate (AB)分子が結合して いた(図 2-21(a))。しかし、L-AAOd/Og では疎水性の芳香環をもつ残基が多く位置し ており、疎水性のポケットを形成していた。特に、crLAAO の 2 番目の AB が結合 する場所には W235 が位置してトンネルの形成を防げている。L-AAOd/Og で見える Aromatic cage は crLAAO では片方がイソロイシンになっているために Cage を形成 しておらず、環を持つ基質(AB)の結合領域を確保していた。一方、Psuedomonas sp. P-501 由来 PAO の場合は、crLAAO より L-AAOd/Og との保存性が高く、多くの残基 が一致していた(図 2-21(b))。しかし、PAO の Leu319 が L-AAOd/Og ではアスパラギ ン酸に変わっており、基質認識部位の上部で基質と相互作用していた。これはリジ ンのような長い基質を認識するために最適化されていると考えられる。 ・基質及び O2 チャンネル O2 分子は L-AAOd/Og の反応が起こるために必要な基質の 1 つである。オキシダ ーゼとして L-AAOd/Og は外部から O2 を取り込んで 2 つの電子を伝達し、H2O2 を生 産する過程を触媒する。またモノオキシゲナーゼとしては、早いスピードで O2 を取 り込み、中間体(C4a-hydroperoxyflavin)を形成する。このように、L-AAOd/Og におい て O2 が外部から導入されるメカニズム及びチャンネル機構は非常に大事であると考 えられる。そこで、本研究では CAVER というプログラムを用いて基質及び O2 のチ ャンネルを見積もってみた。その結果、L-AAOd/Og には Substrate binding domain と Helical domain の間を貫通する 2 つのチャンネルが存在し、そのチャンネルを経てア ミノ酸基質や O2 分子が出入りしていると考えられる(図 2-22(a, b))。 一方、Native L-AAOd/Og の Chain A と Lys 複合体 L-AAOd/Og の Chain A の構造的 類似性を調べたところ、RMSD が約 0.63 Å でほぼ変わりがない構造をとっているこ 110 とを確認した。しかし、Native L-AAOd/Og では欠損していた Substrate binding domain の β16 と α16 を繋ぐフレキシブルなループ(Met419-His428)が基質複合体構造 では見えていた(図 2-23(a))。このフレキシブルなループは基質認識部位から約 15 Å 離れ、チャンネル 2 に隣接していた。そのため、このループがアミノ酸基質または O2 の出入りに関与する可能性があると考え、これらの構造の分子表面図を比べてみ た。その結果、フレキシブルなループは隣の分子との接触面に存在しており、 Native L-AAOd/Og ではループが欠損しているため、両分子の中央が開いている形を していた(図 2-23(a))。また、非対称単位中で四量体として存在する基質複合体の構 造では、四量体のうち 2 分子のみを表示してその裏側を観察して見た。その結果、 フレキシブルなループが固定されて見えていたため、分子表面図では両分子の真ん 中が閉じている形をしていた(図 2-23(b))。 L-AAOd/Og は溶液中で四量体を形成しており(2.11 節結果)、隣に相互作用してい る分子が必ず存在していることを意味する。また、基質が結合してフレキシブルル ープが固定された Closed form になると 2 つのチャンネルのうち、チャンネル 2 が使 用できなくなり、チャンネル 2 を通しては何も出入りができないと考えられる(図 223(b)) 。 こ の よ う に 基 質 の 結 合 に よ っ て チ ャ ン ネ ル が 開 閉 さ れ る 機 構 は 、 LAAOd/Og のオキシダーゼ及びオキシゲナーゼの反応機構を明確にするために重要な 鍵になると考えられる。 ・pCMB 修飾の L-AAOd/Og の構造 L-AAOd/Og の新規構造を解明することと共に本研究の主な目的の 1 つであるモノ オキシゲナーゼからオキシダーゼへの活性変換の構造生物学的メカニズムを明らか にするために、変異体酵素 C254I を用いた構造解析と pCMB 修飾 L-AAOd/Og の構 造解析を行った。しかし、変異体酵素 C254I に関しては、その発現量が極めて少な いため、結晶化スクリーニングを行うことができなかった。Native 酵素の結晶化条 件を用いて結晶化を行ってみたが、結晶は出るものの、沈殿も一緒に沢山出たため、 構造解析に適する結晶を得ることはできなかった。 一方、pCMB 処理実験は、共結晶法とソーキング法を用いて行った。図 2-24 にそ の結果をそれぞれ示した。L-AAOd/Og タンパク質溶液と pCMB を混合して作った共 結晶を用いた構造解析の結果(最大分解能~2.86 Å)(図 2-24(a))、pCMB 処理の構造(左) では FAD が抜けていることが分かった。pCMB 非処理の構造(右)では FAD に該当す る電子マップが強く見えているが、pCMB の処理を受けた構造では FAD に該当する マップが見えなかった。ただし、この現状が本当に pCMB の影響かどうかについて ははっきりは分からないが、他のすべての構造(Native L-AAOd/Og、3 つの基質複合 111 体 L-AAOd/Og の構造)では FAD の結合が見えていたため、pCMB が何らかの作用を 行って FAD の結合を妨げたのではないかと考えられる。 一方、図 2-24(b)にはソーキング法による L-AAOd/Og への pCMB の修飾を試みた 結果を示した(最大分解能~2.8 Å)。pCMB の修飾を受けると水銀からの異常分散が見 えるため、その差フーリエマップを用いて pCMB の修飾を判別した。その結果、LAAOd/Og に含まれる 6 つのシステインのうち、4 つのシステイン(C280, C331, C342, C413)に対しては 3.7 sigma の差フーリエマップで水銀の異常分散が見え、pCMB の 修飾を受けていることが確認できた。しかし、今回の対象残基である C254 を含む 2 つのシステイン残基(C321)では水銀の異常分散が見えず、pCMB による修飾がなか ったと判断される。pCMB 修飾に関しては、C254 の場合分子の奥側に位置している ために修飾されにくいと考えられたが、分子表面に位置している C321 も修飾を受 けなかったことから、単純にシステインの位置が pCMB の修飾を決定する要因では ないと考えられる。 この実験により、pCMB が Cys254 に直接影響を与えていることを証明することは できなかったが、Cys254 の隣に位置する Trp516 の電子マップが非常に弱いことや 共結晶を用いた実験で FAD が抜けた構造をとっていることなどから、pCMB は LAAOd/Og の活性中心構造に何らかの影響を与えており、活性の変換を誘導している と考えられる。 2.15 本章のまとめ Pseudomonas sp. AIU 813 由来の新規二機能性酵素リジンオキシダーゼ/モノオキシ ゲナーゼの立体構造を解明した。本酵素は、L-amino acid oxidase family の特性と flavin monooxygenase family の特性を両方持っており、構造が知られている両酵素の うちでも最も高いアミノ酸配列相同性が 22%前後であるため、分子置換法は適用で きず、Se-Met 置換体を用いた重原子置換法によって立体構造を決定した。ドメイン の構成、各ドメインの構造は既知の LAAO family 酵素らとかなり類似していたが、 基質認識部位に存在する Aromatic cage や D238、R102 などによって、リジンをはじ めとする正電荷を持つ縦に長い基質に特異的に反応する基質結合様式を持っていた。 また、本酵素の分子内に存在する 2 つのチャンネルは、L-AAOd/Og の内部に出入 りする O2 やアミノ酸基質のために存在していることが分かった。Met419-His428 か らなるフレキシブルループは、基質の結合可否によって「Open form」から「Closed form」に Conformational change が行われると考えられる。基質の結合による LAAOd/Og の触媒反応メカニズムを図 2-25 に模式的にまとめた。水溶液中に 4 量体 112 として存在する L-AAOd/Og の内部には十字のトンネルが存在し、そのトンネルを 通して外部から基質が入り込む。その後、各単量体の基質認識部位から 4 量体の中 心部まで通っている「チャンネル 2」を経てアミノ酸基質が分子内に入ってくる。分 子内で基質が結合して FAD が還元状態になるとチャンネル 2 の上部にあるフレキシ ブルループが閉じられ、チャンネル 2 を通しては分子の出入りができなくなる。基 質認識部位から外部の方に通っている「チャンネル 1」を通して O2 が入り、基質認識 部位でオキシダーゼ及びオキシゲナーゼ反応が行われると、FAD は酸化状態になり、 フレキシブルループがまた開く。開いたチャンネル 2 を通して生成物が出て、また 片方のチャンネル 1 からは H2O2(モノオキシゲナーゼの場合は H2O と CO2)が出てく るようなメカニズムになると予測される。 一方、L-AAOd/Og のオキシダーゼ及びオキシゲナーゼ反応の変換機構に関しては、 変異体酵素の発現量が極めて低くなり、タンパク質の溶解度が下がって沈殿しやす くなったことなどの問題で構造解析までは至らなかった。しかし、pCMB を用いた 共結晶やソーキングで得られた情報から pCMB によるシステイン残基の修飾、構造 の変化などを予測することが可能であった。オキシダーゼ及びオキシゲナーゼの選 択的反応機構については、類似酵素である PAO の論文でよく議論されている[25]。 しかし、PAO は異なる 2 つの基質に対して両活性を示しているが(L-Phe にはオキシ ゲナーゼとして、L-Met にはオキシダーゼとして働く)、L-AAOd/Og は 1 つの基質 (L-Lys)に対して両活性を示すことが特徴である。両活性の全般的な反応機構は LAAOd/Og と PAO で大きく変わらないと判断し、PAO の反応機構を基にして LAAOd/Og の反応変換機構について予測した。図 2-26(a)に示した PAO の反応機構に よると、最初に基質が酵素と反応すると脱水素反応(dehydrogenation)が起こって、ヒ ドリド転移により水素原子 1 個が抜けたイミド中間体が作られ、FAD は 2 電子還元 状態になる。その後、酵素がオキシダーゼとして働くか、オキシゲナーゼとして働 くかが決まるが、隣の水分子「wa1」に由来するヒドロキシル基の求核攻撃を受けて、 FAD の酸化とは別個に反応が進むのがオキシダーゼ反応、酸化された FAD の C4aヒドロペルオキシドから酸素原子 1 つを受け取る反応がモノオキシゲナーゼ反応で ある。本研究で明らかにしたイミド中間体構造であるリジン複合体構造を PAO と比 べることで、その反応メカニズムを推測してみた(図 2-26(b))。PAO の構造では wa1 が隣の T313(L-AAOd/Og では T232 に該当)と水素結合をしているが、L-AAOd/Og の 場合 Q258 と結合していた。つまり、L-AAOd/Og のオキシダーゼ反応が起こるため には、Q258 と結合している水分子 wa1 によって求核攻撃をうけて反応が進むと予 測される。それでは、なぜ L-AAOd/Og の C254I の変異体がモノオキシゲナーゼの 活性を失い、オキシダーゼとしての活性のみが上昇するのだろうか? 本研究で解明 した構造と pCMB を導入した構造解析の結果からその理由を推測してみた。まず、 254 番のシステイン残基は基質結合部位の Aromatic cage を構成する W516 の隣に位 置している。そこで、C254I の変異は 516 番目のトリプトファン残基を動かすこと 113 によって基質の結合様式が変わる可能性がある。基質の結合位置は、オキシダーゼ 反応が起こるかオキシゲナーゼ反応が起こるかを決めるうえで非常に重要であると 考えられる。2 つ目の可能性は、pCMB 修飾構造から推測された。今回の pCMB 修 飾構造では C254 には水銀による修飾が観察されなかったことを 2.14 の「pCMB 修飾 の L-AAOd/Og の構造」の結果で述べたが、もし C254 が分子内で大きく動いている と水銀の修飾による異常分散ピークが見えない可能性がある。また、C254 は LAAOd/Og のオキシダーゼ反応を介する水分子 wa1 と水素結合している Q258 とは 4 残基しか離れていない。そこで、C254I の変異体は直接 Q258 に影響を与えて活性を 制御する可能性があると考えられる。実際、C254 と Q258 が含まれるループが pCMB の修飾によって元の位置から約 3~4 Å ほどコンフォメーション変化を起こし ていることが確認できた(図 2-27)。すなわち、ループの位置の変化により、実際に 基質と結合している Q258 と R102 の位置が大きく変わることによって、オキシダー ゼ反応が起こりやすくなる可能性が考えられる。その場合、水分子 wa1 がそのまま の位置で求核攻撃を起こすとは考えにくい。このようなコンフォメーション変化に より、Aromatic cage が部分的に崩れて、水分子が入りやすくなるのではないだろう か。一方、オキシゲナーゼ反応が起こるには、基質が C4a-ヒドロペルオキシドと反 応するために、厳密に正しい位置になければならない。したがって、基質の位置が ずれることによりオキシゲナーゼ反応が起こらなくなり、Aromatic cage の中に入り 込んだ水分子により、オキシダーゼ反応が起こるのではないだろうか。 本研究では、C254I 変異体の構造が得られなかったため、L-AAOd/Og の活性転換 機構を明確に解明することはできなかったが、今回得られた Native L-AAOd/Og およ び基質複合体構造や pCMB 修飾 L-AAOd/Og の構造を通して基質と酸素が利用する 分子内チャンネルの作用機構、モノオキシゲナーゼからオキシダーゼへの活性転換 機構を予測することができた。これらの研究結果は、将来にリジン検出及び定量の ためのリジンバイオセンサーなどの開発を導くことができる意味がある結果だと考 えられる。 114 図表 L-α-AAO Amino acid Pseudomonas AIU 813 (11) L-α-Lys oxidase Trichoderma 100 (5) L-ε-Lys oxidase Marinomonas 100 L-Lysine 100 L-Ornithine 31 18.2 15.1 L-Arginine 6 6.1 < 0.5 L-Phenylalanine 0 8.3 0 L-Tyrosine 0 5.5 0 L-Histidine 0 3.8 0 (6) 「Isobe, K. et al., J. Biosci. Bioeng., 2012. 144, 257-261」より抜粋 表 2-1 3 種類の L-リジンオキシダーゼの基質特異性 L-AAOd/Og は他の L-α-リジンオキシダーゼより高い基質特異性を持っており、L-εリジンオキシダーゼに比べて、広い pH 範囲でリシンに対する活性を示す。 115 Protein (mg) Crude Ni MonoQ Gel-filtration 3921 54.7 37.5 29.4 Total activity (U) 33.7 8.14 5.5 6.8 Specific activity (U/mg) 0.009 0.15 0.15 0.23 Yield (%) Purification (fold) 100 1.4 0.96 0.75 1 16.7 17.0 25.6 表 2-2 L-AAOd/Og の精製表 Ni-chromatography、MonoQ anion-chromatography、Gel filtration chromatography の 3 段 階の精製で目的の蛋白質のみを分離できた。3L の大量培養から最終的に得たタンパ ク量は 29.4 mg、精製率は 25.6 倍であった。 116 表 2-3 X 線回折デー ータ測定と構造 造精密化の統計 計値 117 「Gomezz, D. et. al., B Biochim. Bio ophy. Acta, 2006. 1764, 11577-1585」よ より抜粋 図 22-1 L-リジン-α-オキシ シダーゼ及 及び L-リジン-ε-オキシ シダーゼの の反応スキー ーム L-リジン-α-オキシ シダーゼは は、α-アミ ノ基から起 起こる酸化的脱アミノ ノ化反応に によって が れるが、L-リ リジン-ε-オ オキシダーゼ ゼはその反 反応が ε2-keto-66-aminocapproic acid が生成され アミノ基で起こる るために 2--amino-6-seemialdehydee adipic acid を生成す する。 118 a) b) 図 2-2 L-AAO Od/Og の基 基質と反応産 産物 a) L-AA AOd/Og は L-リジンだ だけではな なく、L-オル ルニチンと L-アルギニ ニンに対す するオ キシダ ダーゼ活性も も有している。 b) 最初 初にオキシダーゼとし して発見され れた L-AA AOd/Og はオ オキシダー ーゼ活性だけ けでは なく、モノオキシ シゲナーゼとしての活 活性も一緒 緒に持つ新規 規二機能性 性酵素である ること が分か かった。 119 「M Matsui. D., et e al., unpubliished data」よ より抜粋 図 2--3 Native L-AAOd/Ogg と変異体酵 酵素 L-AAO Od/Og C2554I の L-リジンオ オキシダー ーゼ/2-モノオキシゲナ ナーゼ活性 性 a) Nativve L-AAOdd/Og, b) 変異体酵素 L L-AAOd/Og g C254I Native L-AAOd/O Og では、モ モノオキシ シゲナーゼ活 活性がオキ キシダーゼ活 活性より 5 倍ほど 高いが、変異体酵 酵素 L-AAO Od/Og C2554I ではオキシダーゼ ゼ活性の方 方が 10 倍以 以上高く なっている。この の結果により、254 番 番目のシステイン残基 基は、L-AA AOd/Og のモノオ の キシゲ ゲナーゼ活性 性からオキ キシダーゼ活 活性への転 転換に大事な役割をし していると と考えら れる。 120 図 2-4 L-A AAOd/Og の分子 子系統樹 121 図 2-5 L-AA AOd/Og の発 発現ベクター Od/Og の大 大腸菌用発現 現ベクター ーpET-15b の構築。富 の 富山県立大学 学の浅野教 教授に提 L-AAO 供していただいた た。 122 図 2-6 L-AAOd/Og L g の X 線回 回折データイ イメージ L-AAO Od/Og の X 線回折デー ータイメー ージ。KEK の PFBL-5A でデータ タ測定を行った。 図 2-7 See-Met 置換 換体 L-AAOd/Og の XA AFS 測定 MAD デ データを測 測定するため め必要な波 波長を XAFS 測定で決 決めた。 123 図 2-8 L-AA AOd/Og の活性測定法 法 AOd/Og の活性測定 の (オキシダー ーゼ活性)の の反応機構 構。基質が酸 酸化すると と生じる a) L-AA 過酸化 化水素(H2O2)をペルキシダーゼと と反応させ せたときの生 生成物であ ある Quino oneimine dye を 555 nm の吸 吸光で測定 定する。 反応で用い いられる試薬 薬の組成 b) a)の反 124 図 2-9 L-AAO Od/Og の大 大量発現·精 精製 段階の精製ス ステップにより L-AA AOd/Og の精 精製を行っ って純粋な目 目的タンパ パク質を a) 3 段 得た。各ステップ プ後のサン ンプルの純度 度を SDS-P PAGE で確 確認した。((ゲル濃度: 10%、 子量マーカ カー、C: 菌体破砕上清 菌 清) M: 分子 b) Gel-ffiltration クロマトグラ ク ラフィー時 時の UV モニ ニターリン ングの結果、 、目的の蛋 蛋白質が 単一ピークとして て検出され れた。 125 図 2-100 ゲルろ過 過による L-A AAOd/Og の 4 次構造 造予測·分子 子量測定 a) 標 準 蛋 白 質 分 子 量 マ ー カ ー の 溶 出 パ タ ー ン 。 分 子 量 マ ー カ ー と し て は 、 Tyrogloobulin、β-aamylase、allbimin、ovvalbumin、ttrypsin の 5 つの標準 準蛋白質を を用いた。 b) マー ーカーと L--AAOd/Og の溶出パタ ターンのプ プロットによ より、Nativve L-AAOd/Og と C254I 変異体の分 分子量を予 予測した。 赤四角が Native L--AAOd/Og 、黄色四角 角が LAAOd/Og C254I 変異体酵素 変 素。 126 図 2-1 11 吸収スペ ペクトル(好 好気条件)の の結果 AOd/Og の吸収スペク クトル。L-A AAOd/Og はフラビン ン酵素であ るため、280 nm a) L-AA でのピーク以外に にも 350 - 500 5 nm で 2 つのピークが観察さ される。 b, c) リ リシンとの反 反応によっ って補因子で であるフラ ラビンが還元 元されるた ため、フラビンの 固有波 波長でのピー ークが減少する。 127 図 2-1 12 吸収スペ ペクトル(嫌 嫌気条件)の の結果 条件下でリシ シンの濃度 度と反応時間 間を変化し しながら酵素の還元度 度を観察し した。高 嫌気条 いリシ シン濃度(6 mM)で長時 m 時間(30 minn)反応させ せたときに、L-AAOd//Og は還元 元状態に なりフラビンの固 固有ピークが見えなく くなった(b))。 128 図 2-13 L L-AAOd/Og の結晶 晶: 0.1 M Soodium acetaate pH 4.6, 8 % Polyethhylene glyco ol 4000 a) Nativve L-AAOdd/Og の結晶 b) Se-M Met L-AAO Od/Og の結晶: 0.02 M Citric acid d, 0.08 M, Bis-Tris B proopane pH 8.8, 8 16% PEG 33350 c) 複合 合体構造のた ため作った た L-AAOd//Og の結晶: 0.15 M DL-Malic D aacid pH 7.0 0, 20 % PEG33550 129 図 2-14 L-A AAOd/Og の全体構造 の 造 a) Ligaand free L--AAOd/Og は、結晶格 格子中二量 量体として存在してい いた。Chaiin A と Chain B を各々緑 緑とシアンで で表示した た。 b) L-A AAOd/Og の基質複合 の 合体構造では は結晶格子 子の中に四量体として て存在して ていた。 2.11 節 節の 4 次構 構造決定の結 結果から水 水溶液中で四 四量体を形 形成していた たため、複 複合体の 四量体 体構造が正し しい構造だ だと考えられ れる。Chaiin A、Chaain B、Chaain C、ChaainD を 各々緑 緑、シアン、ピンク、黄色で示し した。また た、FAD をオレンジで で、基質である LLys を赤 赤のボール ルで表紙した た。 130 図 2-15 L-A AAOd/Og の分子表面図 の 図 量体の分子表 表面図。分 分子の片面に に横長いト トンネルが形 形成されて ている。 a) 二量 b) L-AA AOd/Og の四量体分子 子表面図か からは、分子 子の真ん中を通ってい いる十字の のトンネ ルが存 存在していた た。このトンネルを通 通して基質 質、あるいは は O2 が分子 子内に入り込むと 予測される。 131 図 2-16 L-AAOd/Og の単量体構 構造とトポ ポロジー図 L-AAO Od/Og の単量 量体は FAD D binding ddomain、Su ubstrate binding domaiin、Helical domain の 3 つのドメイン ンからなり、各々のド ドメインを を青、赤、緑 緑で表示し した(色はトポロジ ー図でも同一)。L L-AAOd/Og は 19 本の の α-へリッ ックスと 19 9 本鎖の β--シートで構 構成さ れている。 132 図 2-1 17 DALI serrver による る類似構造の の検索 DALI sserver (http://ekhidna.b biocenter.hellsinki.fi/dalli_server/)を を用いて、L L-AAOd/O Og の類 似構造 造を探索した た。LAAO family に属 属する酵素群が上位に にランクさ れた。 133 図 2-18 L-A AAOd/Og の活性中心 の 心 Od/Og の活性 性中心は、分子の真 ん中に位置 置していた。Aromaticc cage (W516, L-AAO Y473)と と疎水性ポ ポケットが形 形成してお おり、C254 は W516 の隣に存在 の 在していた。シス テイン残基はシア アンで示した。 134 図 2-19 L-AA AOd/Og 基質 質複合体構 構造 基質である L-Lyss、L-Orn、L-Arg が L L-AAOd/Og g と結合し した様子をオ オミットマ マップを 用いて示した。各 各基質の色 色を紫、赤、 オレンジ ジで表示した た。 135 図 2-20 酸化還元状 酸 態に依存す する FAD の構造変化 の a) ①構 構造解析に用 用いた Bp phA4 の結 晶。酸化還 還元状態により結晶の の色が変わ わる。② 単結晶分光学測定 定の結果。③FAD の のイソアロキ キサジン環 環のバタフラ ライ型構造 造変化。 ④二電 電子還元型と と一電子還 還元型ではリ リビチル鎖 鎖が反転して ていること がわかる。 「Senda. M., et al., 化学と生物, 化 2008. 46(100) 689-696」よ より抜粋 b) L-A AAOd/Og の酸化還元 の 状態での FAD の構 構造。本研究 究において ては Native 状態と Lys 複合 合体での FAD FA の構造 造の有意差が が見られな なかった。 136 図 2--21 類似酵 酵素との比較 較 (Stereo view) v AOd/Og と crLAAO (L L-amino aciid oxidase from f Callosselasma Rhoodostoma)の の活性 a) L-AA 中心を重ね合わせ せた。L-AA AOd/Og を緑 緑で、crLA AAO を青で で示した。 L-AAOd/O Og の基 質である L-Lys を赤で、crL を LAAO、PA AO の基質ア アナログで である o-am minobenzoatte をシ アンで表した。 AOd/Og と PAO (L-ph henylalaninne oxidase frrom Psuedo omonas sp. PP-501)の活 活性中心 b) L-AA の比較 較。L-AAOdd/Og を緑で で、PAO を を黄色で示し した。 137 図 2-22 2 L-AAOd/O Og が持つ 2 つのチャ ャンネル a) L-AA AOd/Og の 2 つのチャ ャンネル(ス ステレオ図))。基質結合 合ドメイン ンと Helical ドメイ ンの間を貫通して ていた。これらのチャ ャンネルを を通して基質 質と O2 が分 分子内に入 入り込む と予測される。 b) 2 つの のチャンネ ネルを拡大した図。両 両チャンネルの周りに に疎水性の残 残基が沢山 山位置 していた。 138 図 2-23 2 基質の の結合によ る構造のコ コンフォメーション変 変化 a) Nativve L-AAOdd/Og の構造 造では、Meet419-His42 28 のループ プが欠損し ていた。ル ループ の位置 置はチャンネ ネル 2 の入 入り口と近い いところで である。その の分子表面 面図では、欠 欠損し ているループのた ため、真ん ん中が空けら られていた た。 b) 基質 質結合構造で では Nativee の構造で で欠損したル ループが見 見えていた。 分子表面 面図でも ループ プが見えてい いるため、分子の真ん ん中が埋まっているよ ように見え える。これは は、基 質が結 結合すること とによって構造が Oppen form から Closed form f にコン ンフォメーション 変化をしていると と考えられ れる。 139 図 2-24 pC CMB の修飾 飾を受けた た L-AAOd/O Og の構造 a) 共結 結晶法を用い いた pCMB B 修飾によ る FAD の欠損 の b) ソー ーキング法を用いた pCMB p によ よるシステイ イン残基の の修飾。同じ じ分子内で でも pCMB から修飾を を受けなか かった 2 つの のシステイン(Cys321、Cys254)が が存在した た。波 長 1.00000 Å で測定 定した水銀 銀原子(吸収 収端 1.0093 Å)の異常分 Å 分散差フー リエマップ プをマ ゼンタで示した。 140 図 2-25 基質結 結合による L-AAOd/O Og の触媒反 反応メカニ ズム 基質結 結合の有無に によりコン ンフォメーシ ション変化 化を起こすフ フレキシブ ブルループ((Met419 -His4288)は、基質 質の出入り口 口として作 作用して基質 質や酸素、反応産物な などの移動 動を制御 すると考えられる る。 141 図 2-26 L-AAOd/Og のモノオキ キシゲナー ーゼ/オキシ シダーゼ反 反応の予測 O のオキシダ ダーゼ及び びオキシゲナ ナーゼ反応 応スキーム a) PAO 「K. Ida et al., J. Biocchem., 2011. 150(6), 6599–669)」より抜粋 b) L-AA AOd/Og と PAO の活性中心の重 重ね合わせ せ(ステレオ図)。L-AA AOd/Og(緑))、 PAO(黄 黄) 142 図 2-27 予測 測される L--AAOd/Og の活性転換 換メカニズ ズム より、C254 4 と Q258 が が含まれる るループが元 元の位置か から 3 ~ 4 Å ほど pCMB の修飾によ ョン変化を起こしてい いた。Q258 8 はオキシ シダーゼ反応 応を触媒す するとき コンフォメーショ に関わる水分子と と相互作用する残基で であるため め、この残基 基が含まれ れるループの のコン 変化は L-AAOd/Og の の活性転移機 機構に影響 響を及ぼして ていると考 考えら フォメーション変 合体構造(緑 緑)、pCMB 修 修飾構造(共 共結晶によ よる; 黄) れる。L-Lys 複合 143 引用文献 1. Lukasheva EV & Berezov TT (2002) L-Lysine alpha-oxidase: physicochemical an d biological properties. Biochemistry (Mosc) 67, 1152-1158. 2. Saudubray JM & Rabier D (2007) Biomarkers identified in inborn errors for lysi ne, arginine, and ornithine. J Nutr 137, 1669S-1672S. 3. 山川民夫 (1976) 生化学実験講座11 アミノ酸代謝と生体アミン. 日本生化学会 第1版. 4. Kusakabe H, Kodama K, Machida H, Midorikawa Y, Kuninaka A, Misono H & Soda K (1979) Occurrence of a Novel Enzyme, L-Lysine Oxidase with Anti-Tu mor Activity in Culture Extract of Trichoderma-Viride. Agr Biol Chem Tokyo 43, 337-343. 5. Kusakabe H, Kodama K, Kuninaka A, Yoshino H & Soda K (1980) Effect of L -Lysine Alpha-Oxidase on Growth of Mouse Leukemic-Cells. Agr Biol Chem Tok yo 44, 387-392. 6. Gomez D, Lucas-Elio P, Sanchez-Amat A & Solano F (2006) A novel type of l ysine oxidase: L-lysine-epsilon-oxidase. Biochim Biophys Acta 1764, 1577-1585. 7. Romette JL, Yang JS, Kusakabe H & Thomas D (1983) Enzyme electrode for s pecific determination of L-lysine. Biotechnol Bioeng 25, 2557-2566. 8. Lavagnini MG, Moscone D, Palleschi G, Compagnone D & Cremisini C (1993) Amperometric lysine bioprobes analysis in feeds. Talanta 40, 1301-1306. 9. Kelly SC, O'Connell PJ, O'Sullivan CK & Guilbault GG (2000) Development of an interferent free amperometric biosensor for determination of L-lysine in food. Anal Chim Acta 412, 111-119. 10. Matsuda M & Asano Y (2010) Determination of plasma and serum L-lysine usi ng L-lysine epsilon-oxidase from Marinomonas mediterranea NBRC 103028(T). A nal Biochem 406, 19-23. 11. Isobe K, Sugawara A, Domon H, Fukuta Y & Asano Y (2012) Purification and characterization of an L-amino acid oxidase from Pseudomonas sp. AIU 813. J Biosci Bioeng 114, 257-261. 12. Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, et al. (2005) Complete genome sequence o f the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23, 873-87 8. 13. Ortet P, Barakat M, Lalaouna D, Fochesato S, Barbe V, Vacherie B, Santaella C, Heulin T & Achouak W (2011) Complete genome sequence of a beneficial pla nt root-associated bacterium, Pseudomonas brassicacearum. J Bacteriol 193, 3146. 144 14. Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, Presto n GM, Zhang XX, Moon CD, Gehrig SM, Godfrey SA, et al. (2009) Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluoresce ns. Genome Biol 10, R51. 15. Yamauchi T, Yamamoto S & Hayaishi O (1975) A possible involvement of sulfh ydryl groups in the conversion of lysine monooxygenase to an oxidase. J Biol C hem 250, 7127-7133. 16. Hayaishi O & Hashimoto K (1950) Pyrocatecase - a New Enzyme Catalizing O xidative Breakdown of Pyrocatechin. J Biochem 37, 371-374. 17. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010) PHENIX: a compreh ensive Python-based system for macromolecular structure solution. Acta Crystallo gr D Biol Crystallogr 66, 213-221. 18. Emsley P & Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126-2132. 19. Perrakis A, Morris R & Lamzin VS (1999) Automated protein model building c ombined with iterative structure refinement. Nat Struct Biol 6, 458-463. 20. Vagin A & Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30, 1022-1025. 21. Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, et al. (2012) CAVER 3.0: A Tool for the Ana lysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput Biol 8, e1002708. 22. DeLano WL (2004) Use of PYMOL as a communications tool for molecular sci ence. Abstr Pap Am Chem S 228, U313-U314. 23. Holm L & Sander C (1993) Protein structure comparison by alignment of distan ce matrices. J Mol Biol 233, 123-138. 24. Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S & Vrielink A (200 0) The structure of L-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. Embo J 19, 4204-4215. 25. Ida K, Kurabayashi M, Suguro M, Hiruma Y, Hikima T, Yamomoto M & Suzuk i H (2008) Structural basis of proteolytic activation of L-phenylalanine oxidase f rom Pseudomonas sp. P-501. J Biol Chem 283, 16584-16590. 26. De Colibus L, Li M, Binda C, Lustig A, Edmondson DE & Mattevi A (2005) Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci U S A 102, 12684-12689. 27. Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T & Yokoyama S (2010) Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit h istone demethylase LSD1/KDM1. Biochemistry 49, 6494-6503. 145 28. Krissinel E & Henrick K (2004) Secondary-structure matching (SSM), a new too l for fast protein structure alignment in three dimensions. Acta Crystallogr D Bi ol Crystallogr 60, 2256-2268. 29. Li M, Binda C, Mattevi A & Edmondson DE (2006) Functional role of the "aro matic cage" in human monoamine oxidase B: structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 45, 4775-4784. 30. 千田美紀, 木村成伸, 福田雅夫, 石田哲夫 & 千田俊哉 (2008) 電子伝達タンパ ク質間の酸化還元状態依存的親和性調節機構. 化学と生物 46, 689-696. 31. Faust A, Niefind K, Hummel W & Schomburg D (2007) The structure of a bac terial L-amino acid oxidase from Rhodococcus opacus gives new evidence for th e hydride mechanism for dehydrogenation. J Mol Biol 367, 234-248. 146 総括 147 植物細胞壁は、セルロース、ヘミセルロース、リグニンなどで構成されている。 近年、天然有機物として、セルロースに次いで多量に存在するヘミセルロースの有 効利用が注目されている。ヘミセルロースの主鎖を分解するキシラナーゼ及び側鎖 を分解する様々なアクセサリー酵素については以前から研究が進んでいたが、多く の糖質加水分解酵素の場合、その基質結合サブサイトの-1 位に注目して研究が行わ れていた。本研究では、好熱性細菌 Thermotoga maritima MSB8 由来 α-L-アラビノフ ラノシダーゼ(Tm-AFase)の X 線結晶構造解析を行い、その立体構造を明らかにする ことに成功した(第 1 章)。特に、基質結合様式を解明することにより+側のサブサイ トの基質特異性を同定することが可能になった。サブサイト+側の認識の構造情報は、 糖質関連酵素の糖質合成への利用、難合成オリゴ糖や有用オリゴ糖を高収率で合成 するために非常に重要である。第 1 章の対象酵素である Tm-AFase は、新規合成基 質である DMT 誘導体(DMT-Xyl)と反応して加水分解反応だけではなく、糖重合反応 を触媒することが報告されているため、本研究では Automated docking 法を用いて DMT-Xyl との結合様式を推測してみた。その結果、Tm-AFase は Phe378、Tyr174、 Asp213、Arg256、β-hairpin motif からなる比較的狭い疎水性の基質結合領域(Coin slot-like hydrophobic cavity)を持っており、そのサブサイト+1 側は DMT のような比 較的大きく疎水的な分子と強く結合していることから、Tm-AFase のサブサイト+1 側は DMT 糖を受容するために最適化されていると考えられた。また、本研究では Tm-AFase の立体構造に基づき、Tm-AFase が持つ耐熱性機構の同定を行った。TmAFase は、現在まで知られているアラビノフラノシダーゼの中で最も高い耐熱性を 持つ酵素として報告されている。Tm-AFase は、各々好熱菌、中温菌由来である GsAFase (Geobacillus stearothermophilus 由来のアラビノフラノシダーゼ)と Bl-AFase (Bifidobacterium longum 由来のアラビノフラノシダーゼ)に比べて、好熱性酵素に特 異的なアミノ酸の割合で構成されており、立体構造のコンパクト化、分子内イオン 結合の増加などの耐熱性機構を整えていた。このように Tm-AFase が熱に強い理由 を構造上の情報を用いてしめすことができた。これから好熱性酵素を用いる研究、 あるいは好熱性酵素へのタンパク質エンジニアリングなど様々な分野での応用が可 能であると考えられる。また、DMT 誘導体との結合様式の解析から、DMT 糖を用 いた新たなオリゴ糖合成にも利用されることを期待する。 一方、アミノ酸は単にタンパク質の構成成分として必要であるだけではなく、糖 や脂質の材料としてエネルギー生産、含窒素化合物の合成などに用いられており、 核酸塩基などの重要な構成要素となっている。したがってアミノ酸代謝は生体内で 極めて多様で重要な機能をしているが、ここには沢山のアミノ酸関連酵素が関わっ ている。最近、特定のアミノ酸に高い基質特異性を示すアミノ酸関連酵素を用いた アミノ酸定量キット、検出バイオセンサーなどが医療、食品などの様々な分野にお いて研究開発されている。本研究の第 2 章では、Pseudomonas sp. AIU 813 由来 L-ア ミノ酸オキシダーゼ/オキシゲナーゼ(L-AAOd/Og)の X 線立体構造解析に成功した。 148 L-AAOd/Og はリジンに高い基質特異性を示しており、リジンに対してオキシダーゼ とモノオキシゲナーゼの両方の反応を触媒する新規二機能性酵素である。また、本 酵素に pCMB を添加することによってモノオキシゲナーゼ活性が減少し、オキシダ ーゼ活性が上昇することに着目して、共同研究グループでは変異体酵素 L-AAOd/Og C254I を作製し、pCMB 修飾を受けない高いオキシダーゼ活性のみを持つ酵素を開 発した。本研究が持つ二機能性に注目して、構造既知の LAAO family (L-アミノ酸オ キシダーゼファミリー)と FMO family (フラビンモノオキシゲナーゼファミリー)との アラインメント·分子系統樹解析を行った結果、L-AAOd/Og は両ファミリーとは違 うところに位置していたため、新規のタンパク質ファミリーに属する可能性が示唆 された。この、オキシダーゼとオキシゲナーゼの二機能性を持つ新規ファミリーに ついては現在まで研究されておらず、一部の類似酵素について機能解析が行われて いるが、その構造解析については報告されてない。本研究では、L-AAOd/Og のリガ ンドフリー構造、基質(Lys, Orn, Arg)複合体構造、pCMB 修飾構造を明らかにするこ とにより、L-AAOd/Og の活性転換機構を推測することができた。L-AAOd/Og の活 性中心は、Trp235、Trp418、Phe416 などの芳香環を持つ残基による疎水性ポケット が 形 成 さ れ て お り 、 Trp516 、 Phe473 、 FAD の フ ラ ビ ン 環 に よ っ て 形 成 さ れ た Aromatic cage が基質との結合に重要な役割をしていた。また、活性転換の鍵である と予測される 254 番目のシステイン残基は、Aromatic cage を形成する Trp516 の隣に 位置していたため、この残基に変異が導入されることによって Aromatic cage が影響 を受け、基質の結合様式が変わると予測される。pCMB 修飾の構造からは、C254 と Q258 が含まれるループが元の位置から約 3 ~ 4 Å ほどコンフォメーション変化を起 こしていた。Q258 は L-AAOd/Og がオキシダーゼ反応を触媒するときに関わる水分 子と相互作用をしている残基であるため、この残基が含まれるループのコンフォメ ーション変化は L-AAOd/Og の活性転換機構に大きく影響を及ぼしていると考えら れる。また、本研究では L-AAOd/Og が持つ分子内チャンネルの作用機構について も提案することができた。基質結合の有無によりコンフォメーション変化を起こす フレキシブルループ(Met419-His428)は、基質の出入り口として作用して基質や酸素、 反応産物などの移動を制御すると考えられる。これらの結果は、将来リジン検出及 び定量のためのリジンセンサーなどの開発に繋がると期待される。 上述のように糖質関連酵素及びアミノ酸関連酵素は、食品·医療·エネルギーなど 様々な分野で応用されているために注目される酵素群であり、その立体構造解析は 未来技術への架橋の役割をする基礎研究分野であると考えられる。本研究では、構 造未知の 2 つの酵素の構造上の情報に基づいて DMT 糖の結合様式(第 1 章)、触媒反 応の転換機構·分子内チャンネルの作用機構(第 2 章)などの解析を行い、各基質の認 識·触媒機構を明らかにすることができた。これらの結果から各酵素と基質の相関関 係をより詳しく明らかにしたと考えられ、酵素の基礎研究だけに止まらず、実際に 産業上のレベルまで応用できることを期待する。また、本研究で解決できなかった 149 いくつかの問題点に関しては、これからより更なる研究が必要であると考えられる。 たとえば、第 1 章で用いた DMT 誘導体は難溶性の基質であるため、実際の結晶化 などには使えなかったことから、モデリングだけではなく変異体酵素を用いて構造 解析を行い、本来の構造上の情報を得ることが必要であると考えられる。また、第 2 章で提案した触媒反応の転換機構をより正確に成立するためにも今後 L-AAOd/Og C254I 変異体の構造解析が行われる必要がある。 150 発表論文 本博士論文の第 1 章の内容は、 Bioscience, Biotechnology, and Biochemistry Vol. 76 (No. 2), pp 423–428 (2012) 「 Crystal Structures of Glycoside Hydrolase Family 51 α-L-Arabinofuranosidase from Thermotoga maritima」 (Im, D.-H., Kimura, K., Hayasaka, F., Tanaka, T., Noguchi, M., Kobayashi, M., Shoda, S., Miyazaki, K., Wakagi, T., and Fushinobu, S.) に発表されている。 また、以上の論文は 2012 年 B.B.B.論文賞(日本農芸化学会)を受賞した。 さらに、第 2 章の内容は、 (2013) 「Mutational and crystallographic analysis of L-lysine oxidase/oxygenase from Pseudomonas sp. AIU 813. Interconversion between oxidase and monooxygenase activities」 (Matsui, D.*, Im, D.-H.*, Fukuta, Y., Fushinobu, S., Isobe, K. and Asano, Y.) *Equally contributed (2013) 「Ligand complex structure of L-lysine oxidase/oxygenase and its activity interconversion mechanism」 (Im, D.-H., Matsui, D., Isobe, K., Asano, Y. and Fushinobu, S.) として投稿準備中である。 一方、今回解析した Tm-AFase の構造は PDB (http://www.rcsb.org/pdb/) ID 3UG3 (Ligand free)、3UG4 (Arabinose complex)、3UG5 (Xylose complex)に登録されている。 151 謝辞 本博士論文を完成するまでに、多くの方々のご指導·ご協力を頂きながら、ここま で辿りつくことができました。 私の所属する酵素学研究室において、修士から博士まで5年間研究の指導に当たっ てくださり、細かいところまでサポートしてくださった伏信進矢教授に心から感謝 いたします。いつも素晴らしい助言で研究を支えてくださった若木高善前教授に感 謝いたします。そして、私が日本で勉強できる環境を整えて下さった祥雲弘文元教 授(名誉教授)に感謝いたします。 Tm-AFaseの研究において、Tm-AFaseを提供して下さいました、(独)産業技術総合 研究所の宮崎健太郎博士、ならびに、Xyl-S-DMTおよびDMT誘導体に関するデータ を提供してくださいました、東北大学の正田晋一郎教授、小林厚志助敎に心から感 謝いたします。 L-AAOd/OgのX線結晶構造解析は、岩手大学の礒部公安教授、富山県立大学の浅 野泰久教授、松井大亮博士との共同研究として行われました。貴重な試料や資料、 膨大なデータを提供していただき、円滑に研究を進めることができました。誠にお 世話になりました。また、本研究はJST, ERATOからのご支援を頂きました。 当研究室の同期である伊藤佑さん、宋賢珍さんは、お互いに切磋琢磨しながら実 験を進めることができました。ありがとうございます。その他にも、岩瀬徳康博士、 松岡真生博士、閆震さん、南煐祐さんをはじめとした酵素学研究室の皆様には、実 験のことだけではなく、留学生としての距離感をなくすように協力してくださって 深く感謝しております。また、当研究室のOBである東北大学の日高將文博士、秋田 県立大学の鈴木龍一郎博士、(株)Macrogenの金尚完博士には、細かい実験手法を教 えてくださり、この博士論文に対して多大なる助言をいただきました。ありがとう ございます。 最後に、今までお祈りの背景で自分を支えてくださった家族に感謝して、この博 士論文を締めたいと思います。 2013 年 1 月 152
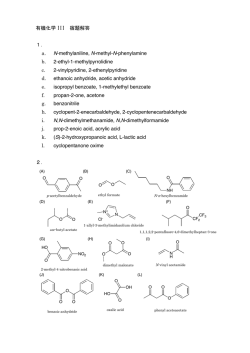
© Copyright 2026